Dette er en veldig stor oversikt over metabolismen fra du spiser til du faster og alt i mellom. I tillegg til ureasyklus. Les denne for å få en oversikt og se alle de røde trådene i metabolismen. Det er både viktig og eksamensrelevant og ha en overordnet kontroll på biokjemien bak metabolismen.
Mettet tilstand
Etter måltid:
Forestill deg at du nettopp har spist et måltid rikt på karbohydrater – kanskje brødskiver, pasta eller ris. I løpet av få minutter begynner glukose å strømme inn i blodet fra tarmen. Konsentrasjonen av glukose i blodplasma stiger merkbart, og dette registreres av β-cellene i de langerhanske øyene i pankreas.
Utskillelse av insulin
β-cellene i pankreas har en spesiell evne til å sense blodsukkeret gjennom GLUT2-transportører, som lar glukose diffundere fritt inn i cellen.
Når glukosekonsentrasjonen i plasma øker, øker også glukoseopptaket i β-cellene – helt passivt. Inne i β-cellen går glukose rett inn i glykolyse og TCA-syklus, noe som øker produksjonen av ATP. Den økte ATP/ADP-ratioen fører til at ATP-sensitive K⁺-kanaler stenges, noe som depolariserer cellemembranen.
Denne depolariseringen åpner spenningsstyrte Ca²⁺-kanaler, og kalsium strømmer inn. Kalsium fungerer som et signalmolekyl og fører til eksocytose av insulinholdige vesikler. Dermed skilles insulin ut i blodet – raskt og i betydelig mengde.
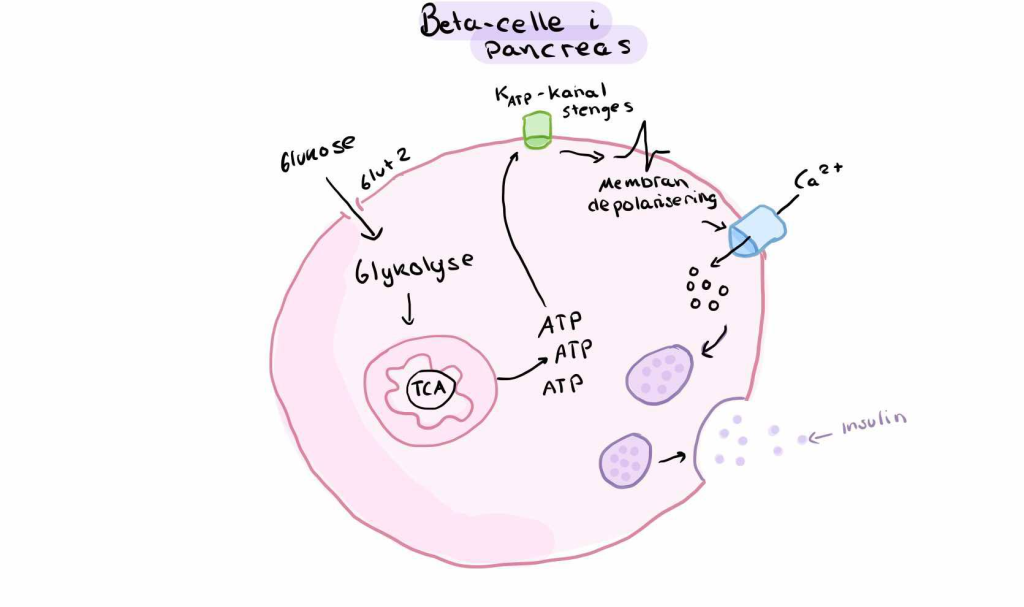
Insulinens systemiske virkninger
Insulin transporteres med blodet og binder seg til insulinreseptorer på målceller, særlig i lever, muskler og fettvev. Disse reseptorene er tyrosinkinase-reseptorer, og aktivering av dem setter i gang en intracellulær signalkaskade. Dette fører til flere avgjørende metabolske endringer, avhengig av celletype:
1. Muskelceller og fettceller: GLUT4-translokasjon
I muskelceller og adipocytter finnes glukosetransportøren GLUT4 lagret i intracellulære vesikler. Når insulin binder seg til sin reseptor, fører det til at disse vesiklene beveger seg mot cellemembranen og fusjonerer med den. Resultatet er en dramatisk økning i glukoseopptaket fra blodet, siden flere GLUT4-transportører nå finnes i membranen.
Dette er en av hovedgrunnene til at insulin senker blodsukkeret etter et måltid.
2. Leverceller: GLUT2 og enzymregulering
Leveren uttrykker GLUT2, en glukosetransportør som alltid er til stede i cellemembranen og ikke reguleres av insulin. GLUT2 har lav affinitet, men høy kapasitet, og tillater passiv transport av glukose etter konsentrasjonsgradienten.
Når insulin er til stede, skjer i stedet reguleringen gjennom modulering av enzymer i glukosemetabolismen. Insulin fremmer:
- Glykolyse (glukoseforbrenning)
- Glykogensyntese (lagring av glukose som glykogen)
- Lipogenese (nydannelse av fettsyrer og fett)
- Og hemmer: glukoneogenese (nysyntese av glukose)
Dette gjør leveren til et sentralt organ for glukosehåndtering: den både bruker, lagrer og omdanner glukose etter behov.
3. Andre vev: Hjernen og erytrocytter
Det er viktig å merke seg at hjernen og røde blodceller ikke trenger insulin for glukoseopptak. Hjernen bruker GLUT1 og GLUT3, som har høy affinitet og gir jevn tilgang til glukose uavhengig av insulinnivå. Røde blodceller har kun anaerob glykolyse og bruker glukose som eneste energikilde – også via GLUT1.
Insulin fungerer altså som en slags metabolsk bryter som setter kroppen i «lagringsmodus». Den øker glukoseopptak, fremmer synteseprosesser og hemmer katabolske veier. Dette skjer raskt etter et måltid, og setter scenen for alt som skal skje videre i metabolismen.
Glykolyse og pyruvatets videre skjebne
Når glukose har kommet inn i cellen – gjennom spesifikke glukosetransportører som GLUT1 eller GLUT4, avhengig av celletype – går det ikke lang tid før den utnyttes til energiproduksjon. Den aller første prosessen i dette omfattende systemet kalles glykolyse.
Dette er en serie av ti enzymkatalyserte reaksjoner som finner sted i cytosol, og som bryter ned glukose – et sekskarbon-sukker – til to molekyler av pyruvat, hver med tre karbonatomer. På veien høstes verdifull energi i form av ATP og NADH.
Et særtrekk ved glykolysen er at den ikke er avhengig av oksygen.
Derfor kan den forsyne celler med energi også i anaerobe omgivelser – noe som er avgjørende for celler uten mitokondrier, slik som røde blodceller, eller i vev som til tider har lav oksygentilførsel, som arbeidende muskulatur under intensiv trening.
Glykolysen kan deles inn i to funksjonelle faser. Den første fasen krever energi og kalles derfor investeringsfasen, hvor to ATP brukes for å «aktivere» glukose.
Den andre kalles høstingsfasen, hvor energien betales tilbake med renter i form av NADH og ATP.
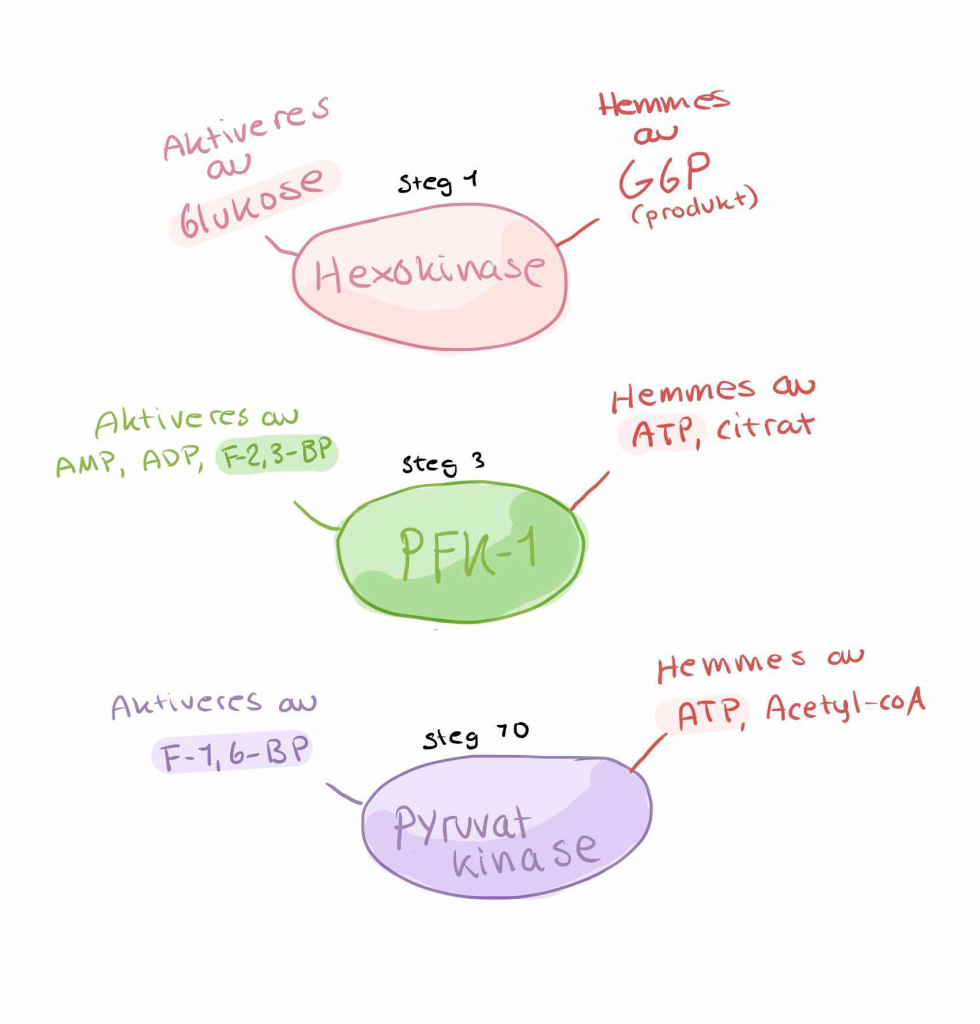
Aller først fosforyleres glukose til glukose-6-fosfat, katalysert av enzymet heksokinase (eller glukokinase i leveren). Denne reaksjonen bruker ett ATP og er irreversibel. Fosforyleringen gjør at glukose fanges inne i cellen, fordi det fosforylerte produktet ikke lenger gjenkjennes av glukosetransportørene og dermed ikke kan diffundere ut igjen. Det er også et første reguleringspunkt – en måte cellen kan bestemme om glukose skal lagres, brukes eller brytes ned.
Deretter omdannes glukose-6-fosfat til fruktose-6-fosfat, som i neste steg fosforyleres igjen, nå av enzymet fosfofruktokinase-1 (PFK-1).
Dette er det viktigste kontrollpunktet i glykolysen.
Her brukes et nytt ATP, og produktet er fruktose-1,6-bisfosfat.
PFK-1 er nøye regulert: enzymet aktiveres av AMP og fruktose-2,6-bisfosfat – signaler på lavt energinivå – og hemmes av ATP, som indikerer at cellen har nok energi. Når glukose når dette trinnet, er løpet kjørt: molekylet er bundet til å gå gjennom resten av glykolysen og ende opp som pyruvat.
Neste fase er en splitting: Fruktose-1,6-bisfosfat spaltes av enzymet aldolase til to trekarbonforbindelser: dihydroksyacetonfosfat (DHAP) og glyseraldehyd-3-fosfat (G3P). Disse to er isomerer, og DHAP konverteres umiddelbart til G3P av enzymet triosefosfatisomerase. Dermed har vi to identiske molekyler av G3P fra hvert glukosemolekyl, og alle de følgende trinnene skjer dermed to ganger per glukose.
I energihøstingsfasen starter vi med oksidasjon. G3P blir oksidert og fosforylert til 1,3-bisfosfoglyserat, samtidig som NAD⁺ reduseres til NADH. Dette er første gang cellen henter ut høyenergi-elektroner i glykolysen, og NADH kan i aerobe celler senere levere disse elektronene til elektrontransportkjeden og bidra til ATP-produksjon.
Neste steg er direkte ATP-produksjon via substratnivåfosforylering: 1,3-bisfosfoglyserat overfører en fosfatgruppe til ADP, og danner dermed ATP og
3-fosfoglyserat. Dette er første gang glykolysen gir ATP tilbake etter investeringen i starten.
3-fosfoglyserat omdannes så videre til 2-fosfoglyserat, og deretter til fosfoenolpyruvat (PEP), et molekyl med en ekstremt høyenergetisk fosfatbinding.
I det siste trinnet av glykolysen overføres denne fosfatgruppen til ADP av enzymet pyruvatkinase, som danner enda et ATP og sluttproduktet pyruvat.
Resultatet etter én runde glykolyse – altså per glukosemolekyl – er:
- Netto +2 ATP (4 produsert, 2 brukt)
- +2 NADH
- +2 pyruvat
Hva som skjer med pyruvat videre, avhenger av oksygentilgangen.
I nærvær av oksygen vil pyruvat transporteres inn i mitokondriene og omdannes til acetyl-CoA av enzymkomplekset pyruvatdehydrogenase, før det går inn i TCA-syklusen. Her kan det videre oksideres til CO₂ og gi langt mer ATP gjennom oksidativ fosforylering.
Men dersom oksygen mangler – for eksempel i anaerob arbeidende muskel – vil pyruvat omdannes til laktat av enzymet laktatdehydrogenase, samtidig som NADH reoksideres til NAD⁺. Dette er nødvendig for at glykolysen skal kunne fortsette, ettersom NAD⁺ må være tilgjengelig i trinn 6. Dermed sørger laktatdannelse ikke bare for å kvitte seg med pyruvat, men også for å opprettholde flyten i hele glykolysen.
Dermed fungerer glykolysen både som startpunkt for aerob energiutvinning, og som en selvstendig nødløsning når oksygen er utilgjengelig – og nettopp dette gjør prosessen så fundamentalt viktig i metabolismen.
Sitronsyresyklus og elektrontransport
Når glykolysen er ferdig og pyruvat har blitt dannet i cytosol, står cellen overfor et avgjørende valg: Skal pyruvat gå videre til fullstendig nedbrytning i mitokondriene, eller skal det reduseres til laktat i cytosol?
I de fleste celler, under aerobe forhold, vil pyruvat transporteres inn i mitokondriematrix, hvor det omdannes til acetyl-CoA.
Dette skjer ved hjelp av et stort multienzymkompleks kalt pyruvatdehydrogenase-komplekset (PDH). Reaksjonen er irreversibel og krever flere koenzymer: TPP, lipoat, CoA, FAD og NAD⁺.
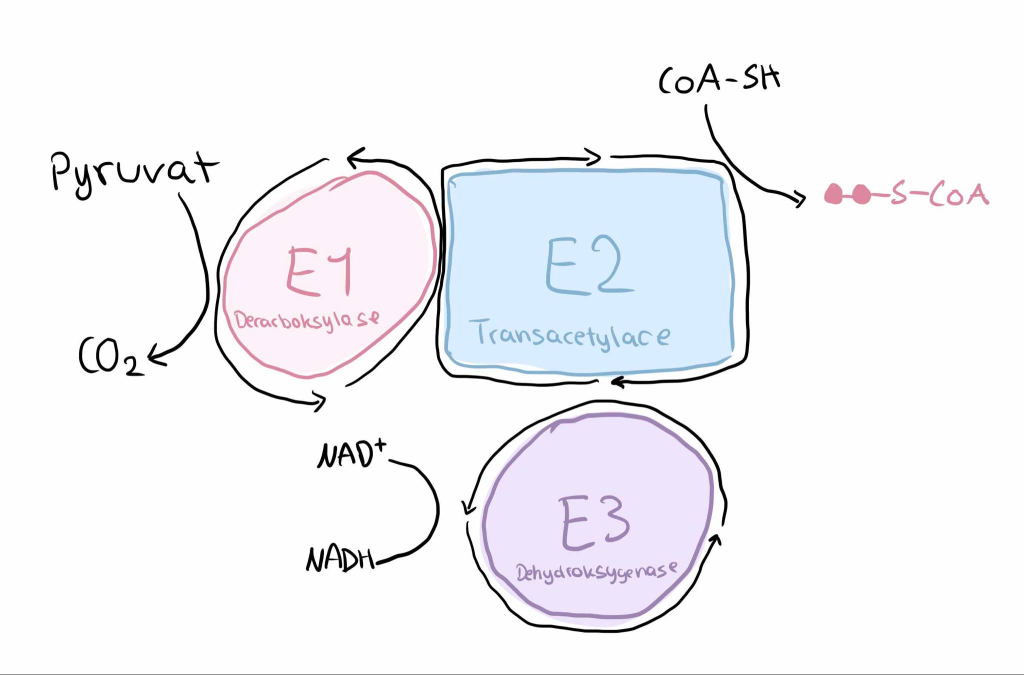
Acetyl-CoA er dermed ikke bare en metabolsk korsvei – det er også inngangsbilletten til sitronsyresyklusen.
Hva skjer om pyruvatdehydrogenase ikke fungerer?
Dersom pyruvatdehydrogenasekomplekset (PDH-komplekset) ikke fungerer, blokkeres koblingen mellom glykolysen og sitronsyresyklusen.
Det betyr at pyruvat ikke kan omdannes til acetyl-CoA, og dermed kan det heller ikke gå inn i mitokondriets TCA-syklus for fullstendig oksidasjon. Som konsekvens vil pyruvat i stedet omdannes til laktat i cytosol, katalysert av enzymet laktatdehydrogenase.
Denne reaksjonen reduserer pyruvat, men samtidig regenererer den NAD⁺, som trengs for at glykolysen skal kunne fortsette. Resultatet blir en økt produksjon av laktat og risiko for laktacidose, særlig i vev med høy glykolytisk aktivitet. Energiutbyttet reduseres dramatisk, ettersom cellene blir avhengige av glykolyse alene for ATP-produksjon.
Sitronsyresyklusen
Inne i mitokondrienes indre kammer, i den såkalte mitokondriematrixen, fortsetter den aerobe nedbrytningen av næringsstoffer med en prosess som er både elegant og grunnleggende for alt liv med oksygenmetabolisme: sitronsyresyklusen. Denne syklusen, også kjent som Krebs-syklusen eller TCA-syklusen (trikarbon-syresyklusen), er et lukket system der karbonatomene i acetyl-CoA fullstendig oksideres, og der cellen fanger opp energien i form av reduserte elektronbærere: NADH og FADH₂. Det er disse molekylene som senere brukes til å generere store mengder ATP i elektrontransportkjeden.
Syklusen begynner med at acetyl-CoA, et tokarbonmolekyl som er felles sluttdestinasjon for både karbohydrater, fett og enkelte aminosyrer, kobles sammen med oksaloacetat, som har fire karbonatomer. Denne kondenseringen danner citrat, en sekskarbonforbindelse, i en reaksjon katalysert av enzymet citratsyntase. Dette er ikke bare første trinn – det er også et kontrollpunkt i syklusen, og gir retning og struktur til prosessen som følger.
Deretter skjer en gradvis omdanning av citrat til isocitrat, og så begynner de energiproduserende reaksjonene for alvor. Enzymet isocitratdehydrogenase katalyserer neste viktige trinn: isocitrat oksideres, og ett karbon spaltes av som CO₂, samtidig som NAD⁺ reduseres til NADH. Dette er første gang i syklusen at karbondioksid frigjøres og høyenergi-elektroner samles inn.
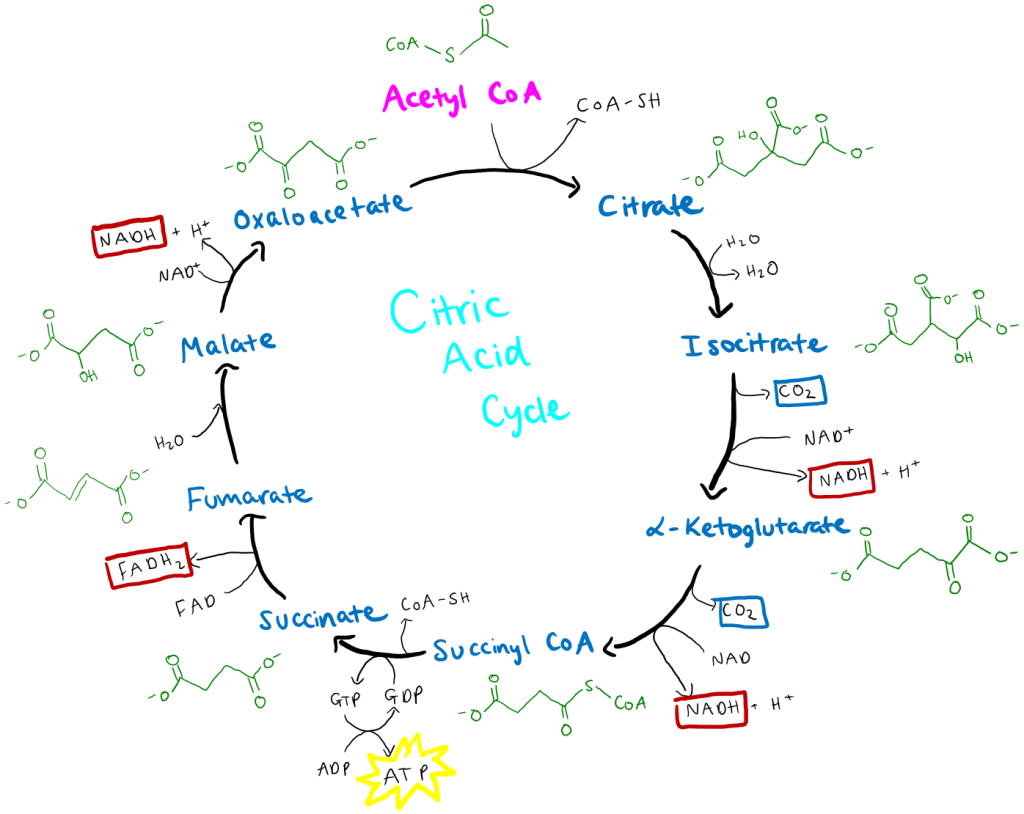
Neste steg er enda kraftigere: α-ketoglutarat, et femkarbonprodukt, gjennomgår en ny oksidativ dekarboksylering katalysert av α-ketoglutaratdehydrogenasekomplekset, et enzymkompleks med mange likheter til pyruvatdehydrogenase. Også her forsvinner ett karbon som CO₂, og det dannes NADH samt et firekarbonmolekyl kalt succinyl-CoA.
Energien fra succinyl-CoA brukes i neste steg til å danne GTP (eller ATP, avhengig av vev) via substratnivåfosforylering, når CoA-gruppen spaltes av og succinat dannes. Nå er vi halvveis, og de fire gjenværende karbonene gjenoppbygges til oksaloacetat i tre trinn, samtidig som mer energi samles opp.
Succinatdehydrogenase, det eneste enzymet i TCA-syklusen som er forankret i den indre mitokondriemembranen, katalyserer oksidasjonen av succinat til fumarat. Dette trinnet er spesielt fordi det danner FADH₂ i stedet for NADH. FADH₂ vil levere sine elektroner direkte inn i elektrontransportkjeden via kompleks II.
Deretter omdannes fumarat til malat, og i det siste energihøstende trinnet oksideres malat til oksaloacetat via enzymet malatdehydrogenase. Her dannes enda et NADH. Nå er vi tilbake der vi startet – og syklusen er klar for en ny runde.
Energiutbytte og funksjon
For hvert molekyl acetyl-CoA som går gjennom TCA-syklusen, produseres det:
- 3 NADH, 1 FADH, 1 GTP (eller ATP), 2 CO₂
Dette betyr at for hvert glukosemolekyl – som gir to acetyl-CoA – får vi dobbelt opp: 6 NADH, 2 FADH₂, 2 ATP/GTP, og 4 CO₂. Alle karbonatomene i glukose er dermed omdannet til gass – og energien er fanget i de reduserte koenzymene.
Det er viktig å forstå at TCA-syklusen ikke primært er en ATP-maskin i seg selv, men en elektron-generator. Dens egentlige oppgave er å produsere så mye NADH og FADH₂ som mulig – fordi det er i elektrontransportkjeden at den virkelig store ATP-produksjonen skjer.
Elektrontransportkjeden – cellens kraftverk
Etter å ha samlet opp store mengder energi i form av NADH og FADH₂ gjennom glykolyse, pyruvatdehydrogenasekomplekset og sitronsyresyklusen, står cellen nå overfor oppgaven å omdanne denne kjemiske energien til brukbar ATP. Det skjer i mitokondrienes indre membran, gjennom et system av proteinmaskiner kjent som elektrontransportkjeden (ETC). Dette er cellens virkelige kraftverk, og her skjer det meste av kroppens ATP-produksjon – drevet av et samspill mellom proteinkomplekser, elektroner, protoner og oksygen.
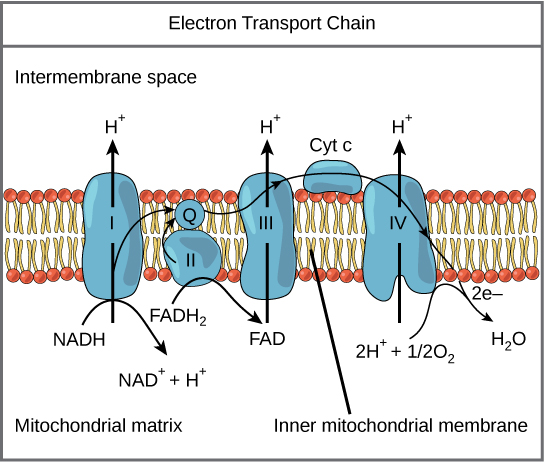
Elektrontransportkjeden består av fire store proteinkomplekser (I–IV) som er integrert i den indre mitokondriemembranen, samt to mobile elektronbærere som fungerer som budbringere mellom dem: ubiquinon (Q) og cytokrom c. Systemet fungerer omtrent som en trapp: elektroner fra NADH og FADH₂ faller energimessig nedover gjennom komplekser med økende elektronegativitet, og denne energiutløsningen brukes til å pumpe protoner (H⁺) fra matrixen ut i det smale intermembranrommet. Resultatet er en kjemisk og elektrisk gradient – en protongradient – som fungerer som et slags oppladet batteri.
Først i kjeden finner vi kompleks I, også kalt NADH-dehydrogenase. Her overføres to elektroner fra NADH til ubiquinon (Q), som dermed reduseres til QH₂. Samtidig pumpes fire protoner fra matrixen ut i intermembranrommet, noe som bidrar kraftig til å bygge opp gradienten. Dette trinnet blokkeres blant annet av giften rotenon, som hemmer elektronflyten fra NADH.
Kompleks II er det samme enzymet som vi kjenner fra TCA-syklusen: succinatdehydrogenase. Her overføres elektroner fra FADH₂ til ubiquinon, men – og dette er viktig – ingen protoner pumpes her. Det betyr at NADH, som går via kompleks I, gir mer ATP enn FADH₂, som hopper over dette første pumpetrinnet.
Neste stasjon er kompleks III, eller cytokrom bc₁-komplekset. Her overføres elektroner fra QH₂ videre til cytokrom c, en liten og mobil elektronbærer som beveger seg på yttersiden av membranen. Under denne prosessen pumpes det ytterligere fire protoner ut, og energien samles videre.
Til slutt ankommer elektronene kompleks IV, også kalt cytokrom c oksidase. Dette er den siste og mest elektronegative komponenten i kjeden. Her overføres elektronene fra cytokrom c til det endelige elektronakseptormolekylet – oksygen (O₂), som dermed reduseres til vann (H₂O). Samtidig pumpes to protoner ut. Dersom oksygen ikke er til stede, stopper hele kjeden opp – noe som forklarer hvorfor celler under anaerobe forhold må ty til laktatproduksjon for å regenerere NAD⁺.
Summen av dette er at cellen bygger opp en kraftig protongradient over den indre mitokondriemembranen. Konsentrasjonen av H⁺ blir mye høyere i intermembranrommet enn i matrixen. Denne gradienten representerer lagret energi – som et vannreservoar bak en demning – og denne energien brukes i neste og siste steg: oksidativ fosforylering.
Oksidativ fosforylering – der protoner blir til ATP
Når elektrontransportkjeden har gjort sitt, har den pumpet store mengder protoner (H⁺) fra mitokondriematrix og ut i intermembranrommet. Denne opphopningen av protoner gir opphav til en protongradient – altså en forskjell i både pH og elektrisk potensial mellom de to sidene av membranen. Dette kalles gjerne for proton-motordrivende kraft (proton motive force). Det er denne kraften som cellen bruker for å drive produksjonen av ATP – gjennom en prosess som kalles oksidativ fosforylering.
Midt i dette står enzymkomplekset ATP-syntase, en fascinerende og høyeffektiv biologisk maskin som sitter innebygget i den indre mitokondriemembranen. ATP-syntase har to hoveddeler: en Fo-del som fungerer som en roterende protonkanal i membranen, og en F₁-del som rager inn i matrixen og faktisk katalyserer dannelsen av ATP. Når protoner strømmer gjennom Fo-delen, settes enzymet bokstavelig talt i rotasjon. Denne mekaniske energien overføres til F₁-enheten, som dermed får riktig konformasjon for å kunne koble sammen ADP og uorganisk fosfat (Pi) til ATP.
For hver runde med protoner som strømmer tilbake, dannes omtrent én ATP per 3–4 protoner. Dette betyr at protongradienten – bygget opp av elektrontransportkjeden – nå omdannes til direkte kjemisk energi i form av ATP. Dette er cellens primære energivaluta, brukt i alt fra muskelsammentrekning og nerveledning til proteinsyntese og cellevekst.
Totalt regner man med at én NADH som leverer elektroner til elektrontransportkjeden, gir opphav til ca. 2,5 ATP, mens én FADH₂ gir ca. 1,5 ATP – dette fordi FADH₂ leverer elektroner senere i kjeden og ikke bidrar til like mange protonpumper.
Klinisk og biokjemisk relevans
Flere stoffer og tilstander kan påvirke denne prosessen dramatisk. Cyanid og karbonmonoksid er dødelige nettopp fordi de hemmer kompleks IV, og hindrer oksygen fra å ta imot elektroner. Når dette skjer, stopper hele kjeden opp, NADH hoper seg opp, og ATP-produksjonen stopper – med katastrofale følger for celler som er avhengige av aerob metabolisme, som hjerneceller.
Andre stoffer, kjent som «uncouplers», som 2,4-dinitrofenol (DNP), gjør membranen lekk for protoner. Da kortsluttes protongradienten, og selv om elektrontransporten fortsetter, kan ikke ATP dannes. Energien går i stedet tapt som varme – en mekanisme kroppen faktisk bruker selv, for eksempel i brunt fettvev, som inneholder proteinet UCP-1 (uncoupling protein-1). Dette gjør at barn og noen dyr kan produsere varme uten å skjelve.
2,4-dinitrofenol (DNP)
2,4-dinitrofenol (DNP) fungerer som en proton-ionofor, som lekker protoner tilbake over den indre mitokondriemembranen uten å gå via ATP-syntase.
Dette fører til at protongradienten kollapser, og cellen mister evnen til å produsere ATP effektivt via oksidativ fosforylering.
Likevel fortsetter elektrontransportkjeden å pumpe protoner, og drivstoff (som fett og glukose) oksideres i høy takt. Men siden energien ikke kan brukes til å lage ATP, frigjøres den i stedet som varme, noe som kan føre til alvorlig temperaturstigning (hypertermi) i kroppen.
Kontroll og regulering av TCA-syklus og elektrontransportkjeden
I cellens stoffskifte er det ikke nok at reaksjonene går riktig – de må også gå i riktig tempo, til riktig tid og på riktig sted. Kroppen har derfor utviklet finstemte reguleringsmekanismer som kontinuerlig tilpasser TCA-syklusen og elektrontransportkjeden (ETC) etter cellens energibehov. Dette skjer både gjennom allosteriske signaler og ved endringer i konsentrasjon av sentrale metabolitter.
Flere av de viktigste enzymene i TCA-syklusen fungerer som reguleringspunkter. Et av disse er citratsyntase, som katalyserer første trinn i syklusen, der acetyl-CoA og oksaloacetat kondenseres til citrat. Dette enzymet hemmes av høye nivåer av ATP og NADH, som signaliserer at energilagrene allerede er fulle. På samme måte hemmes også isocitratdehydrogenase og α-ketoglutaratdehydrogenase, to sentrale enzymer som produserer NADH og fører til CO₂-utskillelse. Når NADH-nivået i mitokondriene blir høyt, fungerer det som en negativ tilbakemelding: det betyr at det allerede finnes nok redusert koenzym tilgjengelig for elektrontransportkjeden, og at det ikke er behov for ytterligere nedbrytning av acetyl-CoA.
Men reguleringen handler ikke bare om bremsing – den handler også om å gire opp når det trengs. Dersom cellen har lavt ATP og høyt nivå av ADP eller AMP, stimuleres både citratsyntase og isocitratdehydrogenase. Dette øker TCA-aktiviteten, slik at mer energi kan høstes fra tilgjengelige substrater. I tillegg har enkelte celler – som muskler – en ekstra regulator: kalsiumioner (Ca²⁺). Under muskelkontraksjon stiger intracellulært Ca²⁺, og dette aktiverer både isocitratdehydrogenase og α-ketoglutaratdehydrogenase. Det er en elegant kobling mellom bevegelse og energifrigjøring: når muskelen trenger å trekke seg sammen, åpnes samtidig energikranene i mitokondriene.
Når det gjelder elektrontransportkjeden, er reguleringen enda mer brutalt enkel: uten oksygen stopper alt. Det er oksygenet som fungerer som den endelige elektronakseptoren i kompleks IV. Dersom oksygen ikke er tilgjengelig, stanses elektronflyten, protongradienten kollapser, ATP-syntesen opphører, og NADH hoper seg opp fordi det ikke lenger kan oksideres tilbake til NAD⁺. Dette skaper raskt et flaskehalsproblem for hele aerob metabolisme – og det er nettopp dette som skjer ved iskemi, altså manglende blodtilførsel. I slike situasjoner må cellen ty til anaerobe løsninger, som glykolyse med laktatdannelse, for å overleve.
Disse reguleringsmekanismene sikrer at cellen verken bruker energi unødvendig, eller går tom for drivstoff når behovet er størst. Metabolismen tilpasser seg – hele tiden, i sanntid – etter kroppens behov.
Lagring av glykogen og fett
Etter et måltid har kroppen ikke bare nok energi til å dekke sine umiddelbare behov – den har mer enn den trenger. Denne energien kan ikke bare fordampe. Den må tas hånd om, struktureres og lagres. Det er her de to viktigste lagringsformene for energi kommer inn i bildet: glykogen og triglyserider. Og igjen er det insulin som styrer prosessen.
Glykogensyntese
Etter et karbohydratrikt måltid strømmer glukose inn i cellene, drevet av insulin. Men kroppen har ikke behov for å bruke all glukosen med en gang. Noe av energien går til umiddelbart forbruk, men overskuddet må lagres – og det skjer blant annet som glykogen, et stort og forgrenet polymer bestående av glukoseenheter. Glykogen fungerer som kroppens hurtigtilgjengelige glukoselager, og er spesielt viktig i lever og skjelettmuskulatur. I muskler fungerer glykogen som et lokalt drivstofflager som raskt kan mobiliseres ved fysisk aktivitet, mens i leveren har det en helt annen funksjon: å opprettholde blodsukkeret mellom måltider ved å frigjøre glukose til sirkulasjonen når det trengs.
Syntesen av glykogen starter med glukose som har kommet inn i cellen via glukosetransportører. Aller først fosforyleres glukose til glukose-6-fosfat (G6P), ved hjelp av enzymet heksokinase i muskelceller eller glukokinase i leverceller. Dette fanger glukosen inne i cellen og gjør den tilgjengelig for videre metabolisme. Deretter skjer en intern omorganisering: enzymet fosfoglukomutase omdanner glukose-6-fosfat til glukose-1-fosfat, som er bedre egnet som utgangspunkt for glykogenoppbygging.
Men for at glukose-1-fosfat skal kunne kobles til et voksende glykogenmolekyl, må det først aktiveres. Dette skjer ved at det kobles til nukleotidet UTP (uridin trifosfat) i en energikrevende reaksjon, og danner UDP-glukose (uridin difosforylglukose) – den direkte donoren av glukoseenheter til glykogen. Denne aktiverte formen fungerer på samme måte som ATP: den bærer både energi og spesifisitet.
Deretter tar enzymet glykogensyntase over. Dette nøkkelenzymet overfører glukoseenheter fra UDP-glukose til en eksisterende glykogenkjede, og skaper α(1→4)-glykosidbindinger mellom glukosemolekylene. Men for at glykogen skal bli biologisk effektivt, må det ikke være en lineær kjede. Det må være forgrenet, slik at det kan mobiliseres raskt. Derfor arbeider et annet enzym parallelt – branching enzyme (også kalt amylo-1,4→1,6-transglukosidase) – og setter inn α(1→6)-glykosidbindinger omtrent hver 8.–12. glukoseenhet. Dette gir glykogen sin karakteristiske struktur med mange «ender», og gjør at glukose raskt kan legges til – eller tas ut – fra mange punkter samtidig.
Hele denne prosessen reguleres nøye av kroppens energistatus og hormoner. Det viktigste signalet for å starte glykogensyntese er insulin, som skilles ut fra bukspyttkjertelen når blodsukkeret er høyt. Insulin aktiverer glykogensyntase via defosforylering, og samtidig hemmer det glykogenfosforylase, enzymet som bryter ned glykogen. Dermed sikres det at cellene bygger opp glykogenlagre når energitilførselen er god, og ikke bryter dem ned samtidig.
Resultatet er en effektiv og dynamisk buffer for blodsukker og muskelenergi. Leverens glykogenlager er typisk brukt opp etter 12–18 timers faste, mens musklene bruker sitt lager under fysisk aktivitet – men ikke deler det med resten av kroppen, fordi de mangler enzymet glukose-6-fosfatase som trengs for å frigi fri glukose til blodet.
De novo lipogenese
Kroppen har en begrenset evne til å lagre glukose som glykogen. Når disse lagrene er fylt opp – i lever og muskler – må overskuddsenergien fra karbohydrater håndteres på en annen måte. I stedet for å kastes bort, omdannes glukose til fett, som deretter kan lagres mer langsiktig i fettvev. Denne prosessen kalles de novo lipogenese, og foregår hovedsakelig i leverceller, men også i mindre grad i fettvev. Prosessen er en omvei, men et elegant svar på kroppens behov for å sikre energireserver til senere bruk.
Alt starter med glukose som, etter å ha blitt brutt ned via glykolyse, danner pyruvat, som igjen omdannes til acetyl-CoA i mitokondriene. Under forhold med høy energitilgang – når ATP og NADH er rikelig – bremser TCA-syklusen opp, og acetyl-CoA akkumuleres. Men acetyl-CoA i seg selv kan ikke krysse mitokondriemembranen. For å komme seg ut i cytosol må det først kobles til oksaloacetat og danne citrat, som deretter transporteres ut via citrat-shuttlen.
I cytosol kløyves citrat igjen til acetyl-CoA og oksaloacetat av enzymet ATP-citrat lyase – en nøkkel i omdannelsen av karbohydrater til fett. Herfra kan fettbyggingen begynne: acetyl-CoA karboksylase (ACC) omdanner acetyl-CoA til malonyl-CoA, som er det regulerte og hastighetsbegrensende trinnet i lipogenesen. Dette enzymet stimuleres av insulin og aktiveres allosterisk av citrat – et klart signal om at kroppen har overskudd av både karbon og energi.
Selve byggingen av fettsyrer skjer gjennom enzymkomplekset fettsyresyntase, som trinnvis kobler sammen acetyl-CoA og malonyl-CoA i en serie reaksjoner som ender med dannelse av palmitat (16:0) – en mettet fettsyre. Hver runde krever også en betydelig mengde NADPH, som leveres av pentosefosfatveien, en alternativ glukosevei som også aktiveres ved høyt energiinntak. Lipogenesen er derfor en syntesevei som krever rikelig med både karbonkilder, energi, og reduktiv kraft.
Når fettsyrene er ferdigsyntetiserte, må de lagres. Dette skjer ved at de bindes til glyserol-3-fosfat for å danne triglyserider (TAGs). I leveren dannes glyserol-3-fosfat enten fra glykolysen (via dihydroksyacetonfosfat) eller direkte fra glyserol gjennom enzymet glyserolkinase. I fettvev finnes derimot ikke dette enzymet, og derfor er fettvev avhengig av glukose for å produsere glyserol-3-fosfat – en viktig forklaring på hvorfor fettlagring i adipocytter krever et karbohydratrikt miljø.
De ferdige triglyseridene pakkes i VLDL-partikler (very low-density lipoprotein) i leveren og sendes ut i blodet. I muskel- og fettvev finnes enzymet lipoproteinlipase (LPL), som spalter triglyseridene og frigjør frie fettsyrer. Disse tas deretter opp av cellene og kan enten forbrennes (særlig i muskel) eller lagres på nytt (særlig i fettvev).
Insulin spiller en sentral rolle også her: det stimulerer LPL i fettvev og dermed øker opptaket av fettsyrer til lagring. Samtidig hemmer det hormonsensitiv lipase (HSL), som normalt ville startet nedbrytning av fett. Dette sikrer at etter et måltid – når energitilgangen er høy – prioriterer kroppen lagring av energi, ikke forbrenning.glyserol via glyserolkinase, men i fettvev må det dannes fra glukose gjennom glykolysen. Dette er grunnen til at fettvev er avhengig av glukose for å kunne lagre fett.
Faste
Tidlig faste:
Når kroppen ikke har fått tilført næring på noen timer, endres de hormonelle signalene som styrer metabolismen.
Etter et måltid dominerer hormonet insulin og fremmer lagring av energi – glukose lagres som glykogen og overskuddsenergi omdannes til fett. Men når fordøyelsen er ferdig og blodsukkeret begynner å falle, synker insulinutskillelsen gradvis.
I stedet stiger nivåene av hormonet glukagon, som skilles ut fra α-cellene i pankreas. Glukagon er kroppens signal om at det er på tide å hente energi fra egne lagre – det stimulerer leveren til å frigjøre glukose og gir kroppen tid til å omstille seg fra en tilstand av energilagring til energimobilisering.
Glukagon virker først og fremst på leverceller.
Når det binder seg til glukagonreseptorer på overflaten av hepatocyttene, aktiveres en G-protein-koblet signalvei som øker nivået av cAMP. Dette fører til aktivering av protein kinase A (PKA), som fosforylerer og regulerer flere sentrale enzymer. Resultatet er en tydelig biokjemisk endring i leverens funksjon: den går fra å være et lagerhus til å bli et produksjonssted for glukose.
To prosesser stimuleres spesielt: glykogenolyse, altså nedbrytning av lagret glykogen, og glukoneogenese, som handler om å lage glukose fra ikke-karbohydratkilder.
Samtidig hemmes glykolyse og glykogensyntese – slik at cellen ikke bruker eller bygger glukose mens den prøver å frigjøre det.
Glykogenolyse: Nedbrytning av glykogen
Den raskeste og mest effektive måten kroppen kan skaffe glukose på i tidlig faste, er ved å hente det fra lagret glykogen i leveren.
Dette lagringsstoffet er en tettpakket form av glukose, bygget opp som en forgrenet polymer med både α(1→4)- og α(1→6)-bindinger.
Nedbrytningen starter med enzymet glykogenfosforylase, som spalter av glukoseenheter i form av glukose-1-fosfat.
Disse omdannes raskt til glukose-6-fosfat, som deretter defosforyleres av enzymet glukose-6-fosfatase – et enzym som kun finnes i lever og nyre.
Den frie glukosen som dannes, kan dermed fraktes ut av levercellen og inn i blodbanen, hvor den bidrar til å opprettholde et stabilt blodsukkernivå.
Det er viktig å merke seg at selv om skjelettmuskulatur også inneholder betydelige mengder glykogen, kan dette ikke brukes til å støtte blodsukkeret. Muskelcellene mangler enzymet glukose-6-fosfatase og kan derfor ikke omdanne glukose-6-fosfat til fri glukose. I stedet bruker musklene glykogenet internt, som energikilde under aktivitet. Muskelglykogen er altså et lokalt, ikke-delbart lager – et privat energireservoar, mens leveren har ansvar for å dekke fellesskapets behov.
Glykogenolyse i leveren er en rask og elegant respons på et fallende blodsukker.
I løpet av minutter etter at glukagon frigjøres, øker glukoseproduksjonen i leveren betydelig. Dette kan dekke kroppens behov for glukose i de første timene og opptil et halvt døgn etter siste måltid. Men leverens glykogenlager er begrenset.
Det kan gi nok glukose i omtrent 12–18 timer, avhengig av hvor mye glykogen som var lagret, og hvor høyt kroppens energibehov er. Når dette lageret begynner å tømmes, må kroppen skaffe glukose på en annen måte.
Allerede før glykogenet er fullstendig brukt opp, starter en gradvis overgang til glukoneogenese – altså produksjon av glukose fra nye kilder.
Dette skjer først og fremst i leveren, men også i nyrebarken under lengre faste. Substratene for glukoneogenesen hentes fra andre vev i kroppen: laktat fra arbeidende muskler og erytrocytter, alanin fra muskelprotein og glyserol fra nedbrytning av fettvev. Leveren setter sammen disse byggesteinene til glukose, som kan sendes ut i blodet og forsyne celler som fortsatt er avhengige av glukose, som hjernen, nyremargen og røde blodceller.
I denne overgangsfasen ser vi også at muskulaturen begynner å bidra med aminosyrer til leveren. Spesielt alanin og glutamin fraktes med blodet til leveren, hvor de omdannes til substrater for glukoneogenese. Samtidig mobiliseres fettvev, og lipolyse aktiveres slik at frie fettsyrer og glyserol frigis. Fettsyrene gir energi til levercellenes egne behov via β-oksidasjon, mens glyserol kan brukes direkte som glukosebygger.
Tidlig faste er derfor en finjustert og dynamisk tilstand hvor kroppen fortsatt prioriterer å holde blodsukkeret oppe, men allerede har begynt å rigge seg til for en mulig sultperiode. Glykogenet brytes ned, men parallelt aktiveres prosesser for mer langsiktig glukoseproduksjon. Hormonsignalene er tydelige og presise, og de metabolske veiene er godt samkjørte – slik at kroppen kan gå mange timer uten mat uten at blodets glukosekonsentrasjon faller farlig lavt.
Bakgrunnen for glukoneogenese:
Når glykogenlagrene i leveren begynner å tømmes, som de typisk gjør innen 12–18 timer etter siste måltid, trenger kroppen en ny og mer bærekraftig løsning for å holde blodsukkeret stabilt. For selv om mange vev i kroppen kan dekke energibehovet sitt ved å forbrenne fettsyrer, finnes det noen vev som er helt avhengige av glukose: hjernen, røde blodceller, nyremarg og deler av retina. Disse vevene mangler mitokondrier eller spesifikke enzymer, og kan ikke bruke fettsyrer eller ketonlegemer effektivt. Det betyr at glukose fortsatt må produseres, selv når det ikke finnes noe igjen å hente fra glykogenlagrene. Løsningen er glukoneogenese – kroppens evne til å produsere glukose fra ikke-karbohydratkilder. Denne prosessen skjer hovedsakelig i leveren, og etter hvert også i nyrebarken ved lengre faste.
Substratene for glukoneogenese: Gammelt blir nytt
Glukoneogenese starter med å samle sammen molekyler som inneholder karbonatomer og som kan omdannes til glukose. De viktigste substratene er:
- Laktat, som dannes i store mengder av erytrocytter (som driver anaerob glykolyse) og arbeidende muskelceller. Laktatet fraktes til leveren og omdannes tilbake til pyruvat, og videre til glukose. Denne sirkelen kalles Cori-syklus.
- Aminosyrer, særlig alanin, som fraktes fra muskelvev til leveren. Alanin omdannes via transaminering til pyruvat, og inngår dermed direkte i glukoneogenesen. Denne prosessen kalles glukose-alanin-syklus.
- Glyserol, som frigjøres når fettvev brytes ned og triglyserider spaltes.
Glyserol omdannes i leveren til glyserol-3-fosfat og deretter til dihydroksyacetonfosfat (DHAP), som er et mellomprodukt i glykolysen og glukoneogenesen.
Fettsyrer i seg selv kan ikke brukes til å danne glukose, siden karbonatomene i acetyl-CoA ikke kan brukes til å bygge opp glukose via reversert TCA-syklus. Men energi fra fettsyreoksidasjon er avgjørende for å drive glukoneogenesen fremover – den gir både ATP og NADH som kreves for å gjøre den energikrevende reaksjonen mulig.
Enzymer og omgåelser: Når glykolysen ikke kan reverseres
Mange av trinnene i glykolysen er reversible og brukes også i glukoneogenesen, men tre av trinnene er irreversible, og må derfor omgås ved hjelp av egne enzymer. Disse tre trinnene i glykolysen, som drives kraftig i én retning, er katalysert av henholdsvis heksokinase, fosfofruktokinase-1 (PFK-1) og pyruvatkinase. Glukoneogenesen må bruke alternative enzymatiske veier for å komme rundt disse “låste” trinnene.
- For å omgå pyruvatkinase, omdannes pyruvat først til oksaloacetat i mitokondriene ved hjelp av pyruvatkarboksylase (et biotin-avhengig enzym). Deretter omdannes oksaloacetat til fosfoenolpyruvat (PEP) av enzymet PEP-karboksykinase (PEPCK). Dette er et viktig regulert trinn.
- For å komme rundt PFK-1, benyttes enzymet fruktose-1,6-bisfosfatase, som hydrolyserer fruktose-1,6-bisfosfat til fruktose-6-fosfat.
- For å omgå heksokinase/glukokinase, bruker leveren enzymet glukose-6-fosfatase, som omdanner glukose-6-fosfat til fri glukose. Dette enzymet finnes kun i leveren og nyrene – og derfor kan bare disse organene bidra direkte til blodsukkeret.
Disse omgåelsene koster energi – faktisk brukes hele 4 ATP, 2 GTP og 2 NADH per glukosemolekyl som dannes fra pyruvat. Det er en energikrevende investering, men en livsviktig en.
Regulering: Glukagon setter retning, insulin stanser
Glukoneogenese er nøye regulert og styres i stor grad av kroppens energistatus og hormonbalanse. Når blodsukkeret faller og glukagon stiger, stimuleres uttrykket og aktiviteten til sentrale glukoneogenetiske enzymer som PEPCK, fruktose-1,6-bisfosfatase og glukose-6-fosfatase. Samtidig hemmes enzymene i glykolysen, slik at de to prosessene ikke går parallelt – en situasjon som ville ført til bortkastet energi (et såkalt “futile cycle”).
Et spesielt og kraftig reguleringspunkt er nivået av fruktose-2,6-bisfosfat (F-2,6-BP), et molekyl som stimulerer glykolyse og hemmer glukoneogenese. Glukagon hemmer dannelsen av F-2,6-BP via aktivering av enzymet FBPase-2, og dermed fremmes glukoneogenese ytterligere.
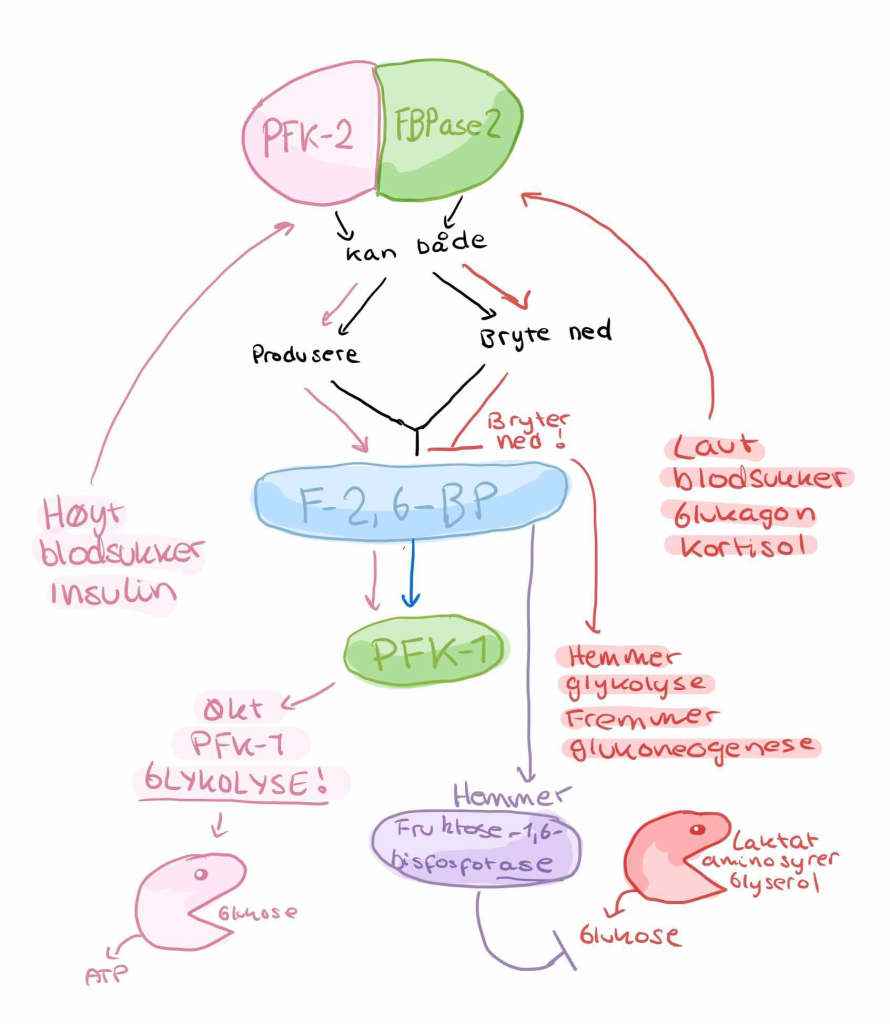
Insulin virker i motsatt retning – det hemmer transkripsjonen av glukoneogenetiske enzymer, øker F-2,6-BP, og aktiverer glykolytiske veier. Dermed blir det hormonelle samspillet mellom glukagon og insulin avgjørende for å bestemme hvilken retning karbonflyten i leveren tar: produksjon eller nedbrytning.
Samspill med andre vev:
Glukoneogenese i leveren støttes kontinuerlig av bidrag fra andre vev. Musklene bryter ned egne proteiner og sender alanin til leveren. Dette skjer spesielt i langvarig faste eller ved stress og sykdom. Fettvevet bryter ned triglyserider til frie fettsyrer, som forbrennes i leveren og gir energien som trengs for glukoneogenesen. Glyserol fra denne nedbrytningen blir også et direkte substrat for glukosesyntese. Erytrocytter og aktive muskler leverer laktat, som gjenbrukes via Cori-syklus.
Denne samhandlingen mellom vev er et strålende eksempel på kroppens integrerte energistyring – hvor hvert vev spiller en rolle, og leveren fungerer som en sentral omkoblingsstasjon for å forvandle restene fra andre prosesser til fersk, sirkulerende glukose.
Langvarig faste og ketogenese
Overgangen til fett: Når glukose blir for dyrt
Når fasten strekker seg utover et døgn, og kroppens glukosebehov fortsatt må dekkes, begynner de hormonelle og metabolske tilpasningene virkelig å tre i kraft. Glykogenlagrene i leveren er nå i praksis tømt, og glukoneogenesen, som tidligere støttet opp glukosebehovet, må nå stå for nesten hele glukosetilførselen. Men glukoneogenese er energikrevende og basert på substrater som kroppen har begrenset tilgang på – særlig muskelproteiner. Hvis kroppen skulle fortsette å hente glukose utelukkende fra aminosyrer, ville det ført til uakseptabel muskelnedbrytning. Derfor må kroppen nå gå over til å bruke en alternativ drivstoffkilde: fettsyrer og ketonlegemer.
I løpet av de første 1–3 døgnene av faste ser vi en markant økning i lipolyse.
Insulin er nå lavt, og hormoner som glukagon, adrenalin og kortisol stimulerer nedbrytningen av triglyserider i fettvev. Resultatet er at store mengder frie fettsyrer og glyserol frigjøres og sendes ut i blodbanen.
Glyserol fraktes til leveren og brukes som substrat i glukoneogenesen, mens fettsyrene tas opp av leverceller og muskelceller og brytes ned gjennom β-oksidasjon i mitokondriene.
For musklene dekker dette stort sett energibehovet.
Men for hjernen og erytrocyttene er ikke fettsyrer et alternativ – de kan ikke krysse blod-hjerne-barrieren, og røde blodceller mangler mitokondrier. Dermed må leveren lage en ny energiform som kan brukes av hjernen: ketonlegemer.
Ketogenese: Når acetyl-CoA går nye veier
I leveren strømmer det nå på med acetyl-CoA fra β-oksidasjonen av fettsyrer.
Under normale forhold ville denne acetyl-CoA gått inn i TCA-syklusen (sitronsyresyklusen), men under faste skjer noe avgjørende: oksaloacetat, som trengs for å kondensere acetyl-CoA med citrat, brukes nå i stor grad til glukoneogenese.
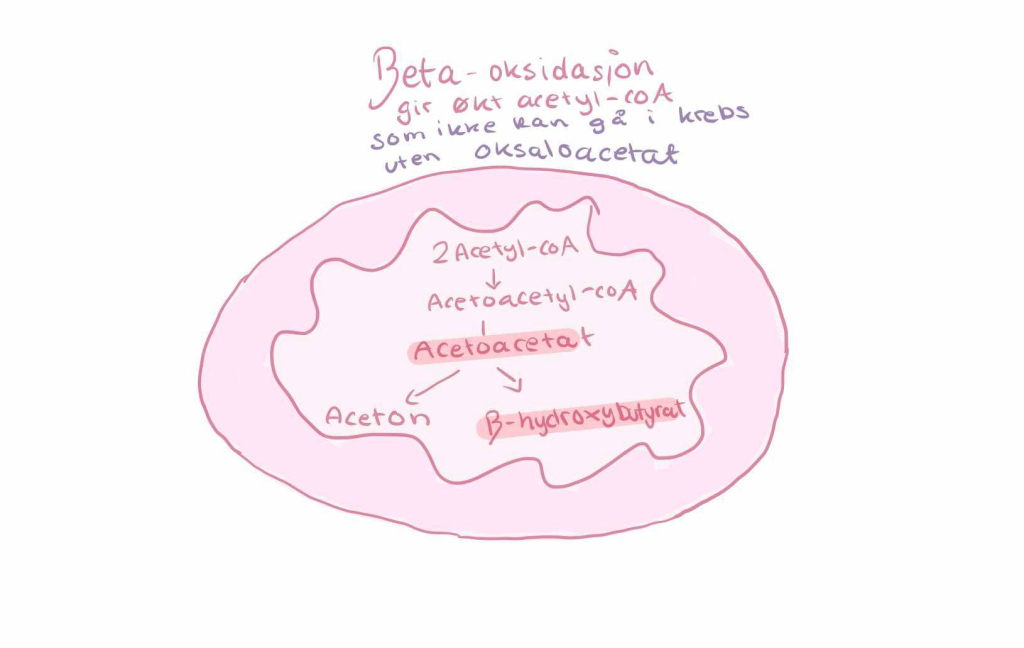
Det betyr at TCA-syklusen ikke går med full kapasitet, og at acetyl-CoA hoper seg opp. Kroppen løser dette ved å bruke overskuddet til å lage ketonlegemer – en ny form for vannløselig drivstoff.
I mitokondriene i levercellene kondenseres to acetyl-CoA til acetoacetyl-CoA, og deretter dannes HMG-CoA (ikke forveksles med kolesterolsyntese i cytosol).
Dette brytes ned til acetoacetat, som enten kan brukes direkte som ketonlegeme, eller omdannes videre til β-hydroksybutyrat, et mer stabilt og energirikt molekyl.
En liten del av acetoacetat kan også spontant dekarboksyleres til aceton, som pustes ut og gir den karakteristiske “fasteånden”.
Ketonlegemer kan fraktes i blodbanen og tas opp av hjernen, hjertet og muskler. I disse vevene omdannes de tilbake til acetyl-CoA og går inn i TCA-syklusen for ATP-produksjon.
Hjernens overgang: Fra glukose til ketonlegemer
Hjernen er et høytenergetisk organ med kontinuerlig behov for energi, og i de første døgnene av faste er den fortsatt helt avhengig av glukose. Men etter 3–5 dager begynner hjernen å tilpasse seg og øke sitt opptak og forbruk av ketonlegemer. Dette er kroppens viktigste sparetiltak: ved å la hjernen bruke ketoner i stedet for glukose, reduseres behovet for glukoneogenese og dermed nedbrytning av kroppens egne proteiner. Faktisk kan hjernen dekke opp til 60–70 % av sitt energibehov med ketonlegemer etter en uke med faste.
Dette er mulig fordi ketonlegemer er små, vannløselige og kan krysse blod-hjerne-barrieren.
Når ketonene er inne i hjernen, omdannes de tilbake til acetyl-CoA, som så går inn i TCA-syklusen (sitronsyresyklusen) for produksjon av ATP – altså energi.
➡️ Unntaket er røde blodceller, som ikke kan bruke ketoner, fordi de mangler mitokondrier.
Det er verdt å merke seg at røde blodceller og nyremarg fortsatt må ha glukose, fordi de ikke har mitokondrier. Det betyr at glukoneogenesen fortsatt må gå, men i langt lavere tempo enn tidligere. Leveren fortsetter derfor å omdanne alanin, glutamin, glyserol og laktat til glukose, men sparer mest mulig på aminosyrene.
Nitrogenbalanse og proteinbesparelse:
I den tidlige fasen av faste ser vi økt nedbrytning av proteiner, særlig fra muskulatur. Det gir leveren substrater til glukoneogenesen, men det gir også en økt utskillelse av urea via nyrene. Etter hvert som kroppen tilpasser seg ketose, reduseres proteinnedbrytningen dramatisk. Dette er en viktig del av overlevelsesstrategien: å spare på kroppens strukturelle proteiner og bremse tapet av muskelmasse. Ketogenesen er derfor ikke bare et energialternativ, men en nitrogenbesparende tilpasning, fordi den gjør at glukoneogenesen kan gå på lavgir.
Oppsummering: En ny metabolsk likevekt
Etter flere døgn uten mat har kroppen nå trappet opp alle de langsiktige, overlevelsesrettede mekanismene. Insulin er lavt, glukagon er høyt, fettsyrenedbrytning er intensivert, ketonlegemer sirkulerer fritt og brukes av hjernen. Glukoneogenesen holdes aktiv, men på et minimumsnivå – nok til å dekke behovene til glukoseavhengige vev, men ikke mer. Denne tilstanden kan kroppen opprettholde i flere uker, forutsatt tilstrekkelig fettreserve og at faste ikke kombineres med alvorlig sykdom. Det er en finjustert og gjennomregulert homeostatisk tilpasning – drevet av en blanding av hormoner, enzymregulering og vevssamarbeid.
Lipolyse og β-oksidasjon: Hvordan fett blir til energi
Fett som energilager:
I motsetning til glykogen, som er lett tilgjengelig men begrenset i mengde, utgjør fettvevet kroppens mest omfattende energireserve. Ett gram fett gir omtrent 9 kcal, mer enn dobbelt så mye som karbohydrater og proteiner. Triglyserider – de lagrede fettmolekylene i fettcellene – består av én glyserol bundet til tre fettsyrer, og disse molekylene kan lagres svært tett, uten vann, og derfor gir de høy energitetthet per gram. Hos en voksen person finnes det flere titalls tusen kilokalorier lagret i fettvev – nok til å overleve i mange uker uten mat.
Men fettlagring og fettnedbrytning er to helt forskjellige tilstander. Når kroppen er i energilagringsmodus – som etter et måltid – stimulerer insulin opptak av glukose og syntese av triglyserider i fettcellene. Når energitilgangen stopper opp, og insulin synker, mens glukagon, adrenalin og kortisol stiger, signaliserer det en ny fase: nå må kroppen bruke av lagrene. Og det starter med lipolyse.
Lipolyse: Når fettvevet frigjør energi
Lipolyse er prosessen hvor triglyserider i fettvev brytes ned til frie fettsyrer og glyserol. Dette skjer i adipocytter – fettcellene – og katalyseres av enzymer som aktiveres av hormonelle signaler. Det første og viktigste enzymet i denne kjeden er hormon-sensitiv lipase (HSL). Når insulin er lavt og glukagon eller adrenalin stimulerer β-adrenerge reseptorer, øker nivåene av cAMP, og dette aktiverer protein kinase A (PKA). PKA fosforylerer og aktiverer HSL, som starter nedbrytningen av triglyseridene.
Lipolyse skjer trinnvis:
- Triglyserider → diglyserider + fettsyre
- Diglyserider → monoglyserider + fettsyre
- Monoglyserider → glyserol + fettsyre
De frie fettsyrene (FFA) som frigjøres, slippes ut i blodet og bindes til albumin for transport. Albumin fungerer som en bærer, fordi fettsyrer er hydrofobe og ikke løselige i plasma alene. Glyserol, derimot, er vannløselig og sendes via blodet til leveren, hvor det kan brukes som substrat i glukoneogenese.
Transport og aktivering av fettsyrer
Før fettsyrer kan brukes som energikilde inne i cellene, må de tas opp og aktiveres. I vev som skjelettmuskel, hjerte og lever, tas fettsyrene opp gjennom spesialiserte transportproteiner, og inne i cytosol bindes de til fettsyre-CoA syntetase (også kalt acyl-CoA syntetase). Dette enzymet katalyserer en ATP-avhengig reaksjon der fettsyren kobles til coenzym A (CoA), og danner acyl-CoA. Dette er det første og nødvendige trinnet i β-oksidasjonen.
Men acyl-CoA kan ikke fritt passere den indre mitokondriemembranen. For å få den inn i mitokondriene, trengs en egen transportmekanisme: karnitin-shuttlen.
Karnitin-shuttlen
For at langkjedede fettsyrer skal kunne brukes som energikilde i kroppen, må de først fraktes dit nedbrytningen faktisk skjer – nemlig inn i mitokondriene. Men her møter de en utfordring: den indre mitokondriemembranen er ugjennomtrengelig for fettsyre-CoA, det aktiverte fettsyremolekylet. Dette er grunnen til at kroppen har utviklet en egen spesialisert transportmekanisme – den såkalte karnitin-shuttlen.
Inne i cytosol, rett utenfor mitokondriet, aktiveres frie fettsyrer ved å binde dem til koenzym A (CoA), i en reaksjon som krever ATP.
Dette danner acyl-CoA. Det aktiverte molekylet kan deretter ikke gå direkte inn i mitokondriet, men må i stedet overføres til karnitin ved hjelp av enzymet CPT-I (karnitinpalmitoyltransferase I), som finnes i den ytre mitokondriemembranen. Resultatet er acyl-karnitin, som kan fraktes over den indre mitokondriemembranen.
Transporten skjer via et spesialisert transportprotein – en translokase – som fungerer som en slags kanal eller port. Når acyl-karnitin er inne i mitokondriematrixen, trer neste enzym inn: CPT-II. Dette enzymet fjerner karnitin og kobler igjen fettsyren til CoA, slik at vi får acyl-CoA tilbake – nå trygt inne i mitokondriet. Karnitinmolekylet sendes samtidig ut igjen til cytosol og kan gjenbrukes. Hele denne prosessen, kjent som karnitin-shuttlen, er avgjørende for at fettsyrenedbrytningen – β-oksidasjonen – i det hele tatt skal kunne finne sted.
Beta-oksidasjon
Når fettsyren endelig er inne i mitokondriene, begynner selve β-oksidasjonen, en systematisk nedbrytning av fettsyrekjeden to og to karbonatomer av gangen. Denne prosessen skjer i en syklus av fire reaksjonstrinn som repeteres til hele kjeden er klippet opp i acetyl-CoA-enheter.
Det første trinnet er en dehydrogenering, der en dobbeltbinding dannes mellom karbon 2 og 3 (alfa og beta) i fettsyrekjeden. Denne reaksjonen katalyseres av acyl-CoA dehydrogenase, og samtidig reduseres FAD til FADH₂, som senere skal bidra til ATP-produksjon i elektrontransportkjeden.
Deretter følger en hydrering, hvor vann tilføres og festes til dobbeltbindingen, slik at det dannes en hydroksylgruppe på beta-karbonet. Så skjer en ny oksidasjon, hvor hydroksylgruppen omdannes til en ketogruppe, og NAD⁺ reduseres til NADH.
Til slutt kommer den viktige thiolytiske spaltningen, hvor molekylet klippes mellom alfa- og beta-karbonet. Dette frigjør én acetyl-CoA, og resten av fettsyren – som nå er to karbon kortere – går rett inn i en ny runde med β-oksidasjon. Prosessen gjentar seg til hele fettsyren er brutt ned.
Et klassisk eksempel er palmitat, en 16-karbon fettsyre. Gjennom 7 runder med β-oksidasjon dannes 8 acetyl-CoA, 7 FADH₂ og 7 NADH. Acetyl-CoA kan så enten gå inn i TCA-syklusen (hvis oksaloacetat er tilgjengelig), eller – i leveren under faste – brukes til å produsere ketonlegemer. FADH₂ og NADH bidrar i elektrontransportkjeden og gir ATP via oksidativ fosforylering.
Totalt gir fullstendig oksidasjon av én palmitat omtrent 106 ATP – en enorm energigevinst, sammenlignet med f.eks. glukose. Derfor utgjør fett kroppens største og mest energirike lager – selv om forbrenningen tar mer tid og krever tilgang på oksygen.
Oppsummert når du faster:
Under faste aktiveres kroppens energimobiliserende systemer. Blodsukkeret faller, insulin synker og glukagon øker – og kroppen går fra å lagre energi til å frigjøre den. Dette fører til tre nøkkelprosesser som henger tett sammen:
For det første starter glukoneogenesen i leveren, hvor kroppen lager ny glukose fra laktat, alanin og glyserol. Denne prosessen er helt nødvendig for å holde blodsukkeret stabilt for vev som fortsatt trenger glukose – som hjerne og røde blodceller. Men glukoneogenesen bruker oksaloacetat som byggestein, og dermed reduseres tilgjengeligheten av oksaloacetat til TCA-syklusen.
For det andre skjer det en omfattende lipolyse i fettvevet, og frie fettsyrer strømmer ut i blodet. Disse tas opp av leveren og muskler og brytes ned via β-oksidasjon til acetyl-CoA – en høyenergisk metabolitt som vanligvis går inn i TCA-syklusen for å gi ATP.
Men her oppstår problemet: Oksaloacetat er nå bundet opp i glukoneogenesen, og TCA-syklusen går på lavgir. Dermed kan acetyl-CoA ikke omdannes til citrat og «bruke» veien sin inn i TCA-syklusen. Det hoper seg opp.
For det tredje løser leveren dette elegant ved å starte ketogenese. Overskuddet av acetyl-CoA omdannes til ketonlegemer (acetoacetat og β-hydroksybutyrat), som fraktes i blodet til hjerne, hjerte og muskulatur, hvor de brukes som alternativt drivstoff. Dette sparer på glukose og minimerer nedbrytning av muskelprotein – en overlevelsesstrategi under lengre faste.
Spesialveier og avfallsutskillelse
Pentosefosfatveien
Når man tenker på hva glukose gjør i kroppen, er det vanlig å tenke på glykolyse, TCA-syklus og ATP-produksjon. Men glukose er ikke bare et drivstoff – det er også et byggemateriale og en kilde til kjemisk kraft som brukes i helt andre sammenhenger enn energiproduksjon. Et tydelig eksempel på dette er pentosefosfatveien – en parallell og helt essensiell metabolsk rute som forsyner kroppen med to viktige ting: NADPH, en sentral reduksjonsfaktor brukt i biosyntese og antioksidantforsvar, og ribose-5-fosfat, byggesteinen i DNA og RNA. Denne veien er ikke en ATP-produsent, og derfor lenge blitt kalt et «biosyntetisk sidespor» – men i virkeligheten er det nettopp denne grenen som gjør cellevekst, steroidhormonsyntese og beskyttelse mot oksidativ skade mulig.
Pentosefosfatveien foregår i cytosol, og starter med det samme molekylet som glykolysen: glukose-6-fosfat. Det er her cellen må velge hvilken vei glukosen skal ta – mot ATP-produksjon, eller mot NADPH og ribose. Hvis cellen har høye energinivåer, men trenger byggesteiner eller reduserende kraft, vil mer av glukose-6-fosfatet kanaliseres inn i denne alternative veien.
Første del av pentosefosfatveien er den såkalte oksidative fasen, hvor NADPH produseres. Her katalyserer enzymet glukose-6-fosfat dehydrogenase (G6PD) den innledende og regulerte reaksjonen, der glukose-6-fosfat oksideres til 6-fosfoglukonolakton, og det første molekylet NADPH dannes. Denne reaksjonen er avgjørende for hele veien videre, og G6PD er derfor nøye regulert – både av NADPH-nivået og cellens behov. I neste trinn oksideres 6-fosfoglukonat videre til ribulose-5-fosfat, samtidig som det andre NADPH-molekylet dannes og én CO₂ spaltes av. Resultatet etter den oksidative fasen er altså: 2 NADPH, 1 ribulose-5-fosfat og 1 CO₂ – alt fra ett glukose-6-fosfat.
NADPH er et kraftig reduksjonsmiddel og brukes i mange forskjellige sammenhenger. I leveren og fettvev er det nødvendig for syntesen av fettsyrer og kolesterol. I binyrebarken og gonadene er det nødvendig for produksjon av steroidhormoner. Og kanskje viktigst av alt: i erytrocytter brukes NADPH til å holde glutation i redusert form. Redusert glutation beskytter celler mot oksidativ skade ved å nøytralisere reaktive oksygenforbindelser. Dette gjør pentosefosfatveien livsviktig for celler som er spesielt utsatt for oksidativt stress – og det forklarer hvorfor G6PD-mangel kan føre til akutt hemolyse hvis kroppen utsettes for infeksjon, medikamenter eller visse matvarer som fava-bønner.
Etter NADPH-produksjonen står cellen igjen med ribulose-5-fosfat, og nå begynner den ikke-oksidative fasen. Her finnes to muligheter, og det er cellens behov som avgjør hvilken vei den velger. Hvis målet er å bygge DNA eller RNA – for eksempel i en celle som skal dele seg – vil ribulose-5-fosfat omdannes til ribose-5-fosfat, en viktig sukkerkomponent i nukleotider. Dette skjer ved en enkel isomerisering, og molekylet kan da brukes direkte i nukleotidsyntesen.
Men hvis cellen ikke trenger å bygge nye nukleinsyrer, vil ribulose-5-fosfat i stedet gå inn i en serie reversible reaksjoner katalysert av enzymene transketolase og transaldolase. Disse enzymene flytter to- og trekarbonfragmenter mellom ulike sukkermolekyler og danner fruktose-6-fosfat og glyseraldehyd-3-fosfat – to mellomprodukter fra glykolysen. På denne måten kan karbonet fra glukose komme tilbake inn i hovedstrømmen av metabolismen, slik at ingenting går til spille.
Denne fleksibiliteten gjør at pentosefosfatveien kan tilpasses enhver situasjon. Hvis cellen trenger kun NADPH, men verken ATP eller nukleotider, kan hele glukosemolekylet kjøres gjennom den oksidative fasen og sukkerrestene resirkuleres tilbake inn i glykolysen. Hvis cellen trenger ribose-5-fosfat, men ikke NADPH, kan de glykolytiske mellomproduktene (fruktose-6-fosfat og glyseraldehyd-3-fosfat) omdannes baklengs til ribose. Og hvis den trenger både NADPH og ribose, kjøres begge faser av pentosefosfatveien sammenhengende.
Kort oppsummert er pentosefosfatveien ikke en energiproduserende vei, men en biosyntetisk og beskyttende mekanisme. Den er aktiv i lever, fettvev, binyrebark, gonader, melkekjertler, erytrocytter og andre vev der syntese eller redoksbalanse er viktig. Den er også et av de beste eksemplene på hvordan kroppen kan ta et og samme utgangspunkt – glukose – og styre det i helt ulike retninger ut fra behov.
Ureasyklus
I det store, koordinerte nettverket av metabolismen står cellene ikke bare overfor utfordringen med å skaffe energi, men også med å kvitte seg med avfall. Spesielt krevende er håndteringen av nitrogen, som frigjøres når aminosyrer brytes ned. Hver gang en aminosyre deamineres – altså mister sin aminogruppe – dannes ammoniakk (NH₃), et stoff som er både giftig og vannløselig. Fordi selv små mengder ammoniakk i blodet kan være nevrotoksisk, må kroppen sørge for en trygg og effektiv metode for å eliminere det. Løsningen er ureasyklusen – en elegant og livsnødvendig biokjemisk syklus som foregår i hepatocyttenes mitokondrier og cytosol, og som gjør om ammoniakk til urea, et trygt, vannløselig og lett utskillelig molekyl som skilles ut med urinen.
Ureasyklusen er tett koblet til proteinmetabolismen. Når proteiner brytes ned i tarmen, eller når aminosyrer brukes som energikilde i vev, fraktes nitrogen til leveren i form av alanin og glutamin – to ikke-toksiske transportformer for aminogrupper. I leveren gjennomgår disse transaminering, og ammoniakk frigjøres. Nå må ammoniakken raskt inn i ureasyklusen.
Syklusen starter i mitokondriet, hvor enzymet karbamoylfosfatsyntetase I (CPS I) katalyserer første trinn: ammoniakk (NH₃) og karbondioksid (CO₂) kobles sammen til karbamoylfosfat. Dette trinnet krever to ATP og er det hastighetsbegrensende trinnet i hele syklusen. CPS I aktiveres av N-acetylglutamat, som fungerer som en signalgiver for at det er overskudd av nitrogen i cellen.
Karbamoylfosfat går deretter inn i en kondensasjonsreaksjon med ornitin – et ikke-proteinogent aminosyrederivat – og danner citrullin, katalysert av enzymet ornitin transkarbamoylase. Citrullin transporteres ut av mitokondriet og inn i cytosol, hvor resten av syklusen fortsetter.
I cytosol skjer nå en ny viktig koblingsreaksjon: citrullin reagerer med aspartat, som kommer fra transaminering av oksaloacetat, og sammen danner de argininosuccinat, katalysert av argininosuccinat syntetase. Denne reaksjonen bruker ytterligere én ATP (men i form av energirikt AMP, så det tilsvarer to ATP). Her ser vi at aspartat fungerer som donor for det andre nitrogenatomet i urea, og koblingen mellom aminosyremetabolisme og ureasyklus blir tydelig.
Argininosuccinat spaltes i neste trinn til arginin og fumarat. Fumarat går tilbake til mitokondriene og kan inngå i TCA-syklusen – et vakkert eksempel på hvordan nitrogenavfall og karbonmetabolisme er integrert. Arginin går videre til siste trinn, hvor enzymet arginase spalter det til urea og regenererer ornitin, som går tilbake inn i mitokondriet for å starte syklusen på nytt.
Urea, som nå er ferdigsyntetisert, fraktes ut i blodbanen og skilles ut via nyrene. På denne måten blir to nitrogenatomer – ett fra ammoniakk og ett fra aspartat – effektivt og trygt eliminert fra kroppen.
Energiforbruk og betydning
Ureasyklusen krever energi: fire ATP per ureamolekyl. Det kan høres mye ut, men det er en nødvendig investering for å unngå opphopning av toksisk ammoniakk. Samtidig genererer tilknytningen til TCA-syklusen – via fumarat og aspartat – en slags metabolsk kobling mellom nitrogenhåndtering og karbonstoffskiftet. Dette koblingssystemet kalles ofte for aspartat-argininosuccinat shuttlen.
Klinisk relevans
Dysfunksjon i ureasyklusen kan ha alvorlige konsekvenser. Medfødte defekter i enzymer som CPS I eller ornitin transkarbamoylase fører til hyperammonemi, hvor ammoniakk hoper seg opp i blodet. Dette kan gi encefalopati, sløvhet, oppkast, og i verste fall koma – spesielt hos nyfødte. Behandlingen involverer spesialdietter med redusert proteininntak, samt medisiner som binder nitrogen og fremmer alternativ utskillelse.
Koblingen mellom ureasyklus og TCA-syklusen: En metabolsk dialog
Ureasyklusen og sitronsyresyklusen (TCA-syklusen) er to svært ulike, men likevel nært forbundne metaboliske prosesser. Den ene har som hovedformål å kvitte seg med nitrogenavfall, mens den andre har som oppgave å hente ut energi fra karbonkjeder. Det er lett å tenke på dem som to separate veier, men i virkeligheten snakker de med hverandre – via en biokjemisk bro som gjør stoffskiftet effektivt, koordinert og økonomisk. Denne broen kalles gjerne aspartat–argininosuccinat-shuttlen.
Det starter med et spørsmål: Hvor kommer nitrogenatom nummer to i urea fra? Svaret er aspartat – en ikke-essensiell aminosyre som dannes i TCA-syklusen ved transaminering av oksaloacetat. Dette skjer i levercellene, hvor TCA og ureasyklus kjører parallelt, og substratene kan flyte mellom de to prosessene. Aspartat transporteres fra TCA-syklusens intermediater til cytosol, hvor det kondenseres med citrullin for å danne argininosuccinat – et mellomtrinn i ureasyklusen.
Deretter følger neste nøkkelpunkt: Når argininosuccinat spaltes av enzymet argininosuccinat lyase, får vi arginin (som gir opphav til urea) og fumarat – og nettopp her oppstår forbindelsen tilbake til TCA-syklusen. Fumarat, som er et firekarbonintermediat, transporteres fra cytosol tilbake inn i mitokondriene og går rett inn i TCA-syklusen igjen. Her kan det enten omdannes til malat, og videre til oksaloacetat, eller brukes til regenerering av aspartat via transaminering.
Denne kontinuerlige syklusen – der aspartat går inn i ureasyklusen og fumarat kommer ut og returneres til TCA – gjør at nitrogenutskillelsen og energimetabolismen er tett integrert. Karbonrammene fra aminosyrer utnyttes til energi, mens nitrogen skilles ut. Energi fra TCA-syklusen (via NADH og ATP) brukes til å drive ureasyklusens energikrevende trinn, og metabolitter resirkuleres elegant mellom de to.
Denne sammenkoblingen har også klinisk betydning. Ved leversvikt eller enzymdefekter som påvirker ureasyklusen, kan opphopning av ammoniakk og forstyrret TCA-syklus føre til alvorlige metabolske kriser. Det understreker hvor dypt integrert nitrogen- og karbonmetabolismen er – og hvor viktig leverens rolle er i å balansere begge.
Oppsummering
Tenk deg kroppen som et logistikksystem, hvor hvert molekyl har en vei å gå, en oppgave å fylle og en dør å velge. Alt begynner med glukose, som slippes inn i cellene ved hjelp av glukosetransportører – GLUT1 og GLUT3 i hjernen, GLUT2 i leveren, GLUT4 i muskler og fettvev, aktivert av insulin. Inne i cellen er glukose som et råmateriale på lageret. Enten brukes det med en gang, eller lagres for senere. Ved høyt energinivå lagres glukose som glykogen via glykogensyntese, eller sendes inn i pentosefosfatveien for å lage NADPH og ribose-5-fosfat. Men om cellen trenger energi, slippes glukosen inn i glykolysen, hvor den kuttes i to og danner pyruvat, og på veien fanges noe energi i form av ATP og NADH.
Pyruvat står så ved et avgjørende veikryss. I nærvær av oksygen og fungerende mitokondrier, transporteres pyruvat inn og omdannes til acetyl-CoA, et slags knutepunkt i metabolismen. Acetyl-CoA kan gå videre til TCA-syklusen, hvor det reagerer med oksaloacetat og gjennomgår en serie dekarboksyleringer og oksidasjoner. Her frigjøres CO₂ som avfall, og samtidig lages store mengder NADH og FADH₂, som er som ladde batterier. Disse sendes til elektrontransportkjeden, hvor elektronene deres brukes til å pumpe protoner og bygge opp en protongradient. Når protonene strømmer tilbake gjennom ATP-syntase, genereres den største energigevinsten: oksidativ fosforylering.
Men acetyl-CoA har flere dører. Ved overskudd på energi, særlig etter et karbohydratrikt måltid, blir det ikke brukt i TCA-syklusen, men eksporteres som citrat til cytosol, hvor det omdannes tilbake til acetyl-CoA og brukes til å bygge fettsyrer i de novo lipogenese. Disse fettsyrene kobles til glyserol-3-fosfat og pakkes som triglyserider, som sendes ut som VLDL eller lagres i fettvev. Fett er kroppens langsiktige energibuffer.
I en motsatt situasjon – under faste eller sult – stenger insulinportene, og glukagon åpner dører til mobilisering. Glykogen brytes ned (glykogenolyse), og glukoneogenese starter fra substrater som alanin, laktat og glyserol. Men samtidig mobiliseres fettlagrene. Lipolyse frigjør fettsyrer og glyserol. Fettsyrene transporteres til mitokondriene via karnitin-shuttlen og gjennomgår β-oksidasjon, som kutter dem i to-karbonenheter og lager mer acetyl-CoA. Nå er TCA-syklusen undertrykt fordi oksaloacetat trekkes til glukoneogenese, og overskuddet av acetyl-CoA går inn i ketogenese, hvor ketonlegemer dannes. Disse fraktes i blodet til hjernen, som i lengre faste bruker dem som alternativ drivstoff.
Underveis håndterer kroppen avfallsstoffer. Ammoniakk fra aminosyrekatabolisme går inn i ureasyklusen, og CO₂ fra TCA-syklusen pustes ut. Protoner pumpet ut i elektrontransportkjeden brukes til å drive ATP-syntese, og overskudd av nitrogen skilles ut via nyrene. Energi er hentet ut, biprodukter håndtert, og nye byggesteiner er klare for neste runde.
📚 Anki-kort
Obs, tomt! Kommer etterhvert <3
📝 Eksamensoppgaver
Obs, tomt! Kommer etterhvert <3
👨⚕️ Klinisk case
Obs, tomt! Kommer etterhvert <3
❓ Test deg selv
Obs, tomt! Kommer etterhvert <3