Innledning
Metabolismen av heme er en grunnleggende biologisk prosess som kobler sammen blodets funksjon, leverens avgiftningssystem og kroppens avfallshåndtering. Heme er en kompleks, ringformet struktur som inneholder jern, og finnes i en rekke viktige proteiner i kroppen, deriblant hemoglobin, myoglobin og flere enzymer involvert i cellenes energiomsetning. Uten heme ville ikke oksygentransport, energiproduksjon eller mange av kroppens forsvarsmekanismer vært mulig.
Men heme er ikke bare nyttig; det er også potensielt farlig. Frie hemegrupper og fritt jern kan fremme dannelse av reaktive oksygenforbindelser som skader celler og vev. Kroppen har derfor utviklet et nøye regulert system for å produsere, håndtere og bryte ned heme på en trygg måte.
Denne prosessen starter med biosyntesen av heme, som skjer primært i benmargen og leveren. Når heme har gjort sin nytte, må det brytes ned kontrollert, slik at det ikke forårsaker skade. Nedbrytningen fører til dannelse av bilirubin, et fargestoff som videre behandles i leveren og skilles ut gjennom galle og tarm. Langs denne reisen spiller leverceller, blodceller, makrofager og tarmbakterier hver sin avgjørende rolle.
Metabolismen av heme har også stor klinisk betydning. Forstyrrelser i hemesyntesen kan gi opphav til sjeldne, men alvorlige sykdommer som porfyri. Svikt i nedbrytningen eller utskillelsen av bilirubin kan føre til gulsott (ikterus), et synlig tegn på underliggende lever- eller blodsykdom.
I denne gjennomgangen skal vi følge heme gjennom hele livssyklusen – fra byggingen i cellens små fabrikker, via transport og funksjon, til nedbrytningen og utskillelsen. Samtidig vil vi belyse de viktigste medisinske tilstandene som oppstår når denne balansen forstyrres.
Hemeproteiner
Heme er en liten, men helt essensiell komponent i mange av kroppens viktigste proteiner. Et hemeprotein er et protein som inneholder én eller flere hemegrupper som en integrert del av sin struktur. Disse hemegruppene fungerer ofte som aktive sentre hvor viktige kjemiske prosesser finner sted – for eksempel oksygentransport, elektronoverføring og nedbrytning av skadelige forbindelser.
Hva er en hemegruppe?
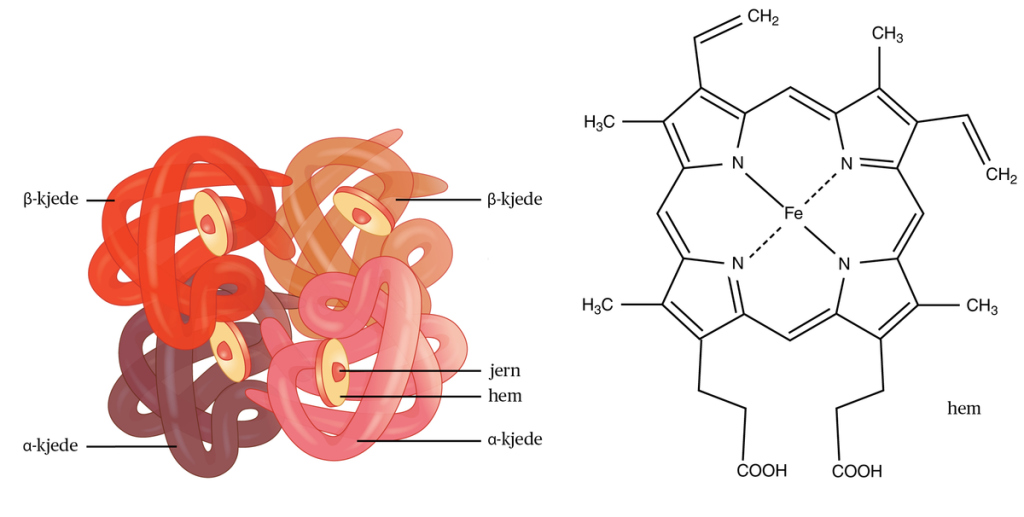
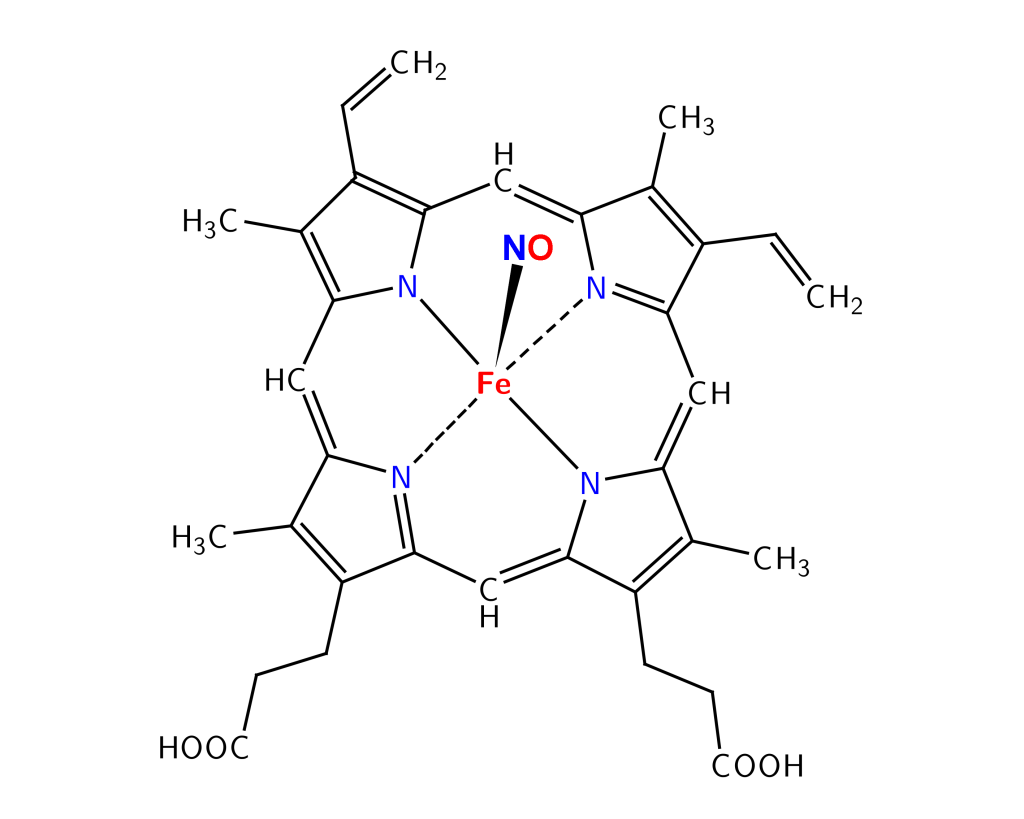
En hemegruppe består av en ringformet struktur kalt en porfyrinring, bygget opp av fire sammenkoblede pyrrolringer. I midten av ringen sitter et jernatom (Fe²⁺ eller Fe³⁺) som kan binde, frigi eller overføre elektroner eller små molekyler som oksygen (O₂), avhengig av hvilken type hemeprotein det er en del av.
Selve porfyrinringen gir heme sin karakteristiske rødfarge, som vi kjenner fra blod.
Oppbygning av et hemeprotein
I et hemeprotein er hemegruppen ikke bare fysisk bundet inn i proteinet; den er også funksjonelt avgjørende. Proteinet rundt hemegruppen – kalt globindelen – former en spesialisert lomme som beskytter heme og kontrollerer hvilke molekyler som kan binde seg til jernet.
Slik sørger proteinet for at heme kan utføre sin spesifikke rolle på en trygg og kontrollert måte.
Eksempler på viktige hemeproteiner
Flere sentrale proteiner i kroppen vår er hemeproteiner:
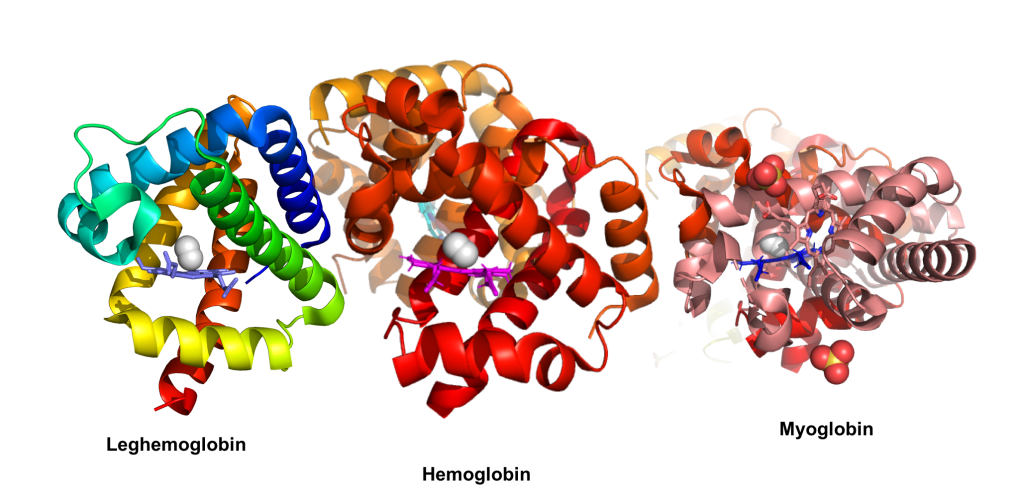
- Hemoglobin: Finnes i røde blodceller og binder oksygen i lungene for transport til kroppens vev. Hemoglobin består av fire polypeptidkjeder, hver med sin egen hemegruppe, slik at ett hemoglobinmolekyl kan bære fire oksygenmolekyler samtidig.
- Myoglobin: Ligner på hemoglobin, men finnes i muskelceller hvor det fungerer som et oksygenlager som kan frigjøre oksygen under perioder med høy belastning eller lav oksygentilførsel.
- Cytokromer: Er hemeholdige proteiner som spiller en sentral rolle i cellenes elektrontransportkjede, hvor de hjelper til med å produsere ATP i mitokondriene.
- Katalase: Er et enzym som beskytter cellen mot oksidativ skade ved å bryte ned hydrogenperoksid (H₂O₂) til vann og oksygen.
Hver av disse proteinene utnytter hemegruppen på litt forskjellige måter, men fellestrekket er at jernet i heme fungerer som et aktivt senter for å binde, frakte eller overføre kjemiske forbindelser.
Betydningen av hemeproteiner
Hemeproteiner er avgjørende for liv. Uten hemoglobin ville ikke oksygen kunne fraktes effektivt fra lungene til vevene. Uten cytokromer ville cellene våre ikke kunne produsere energi. Og uten katalase ville kroppen være utsatt for alvorlig skade fra frie radikaler.
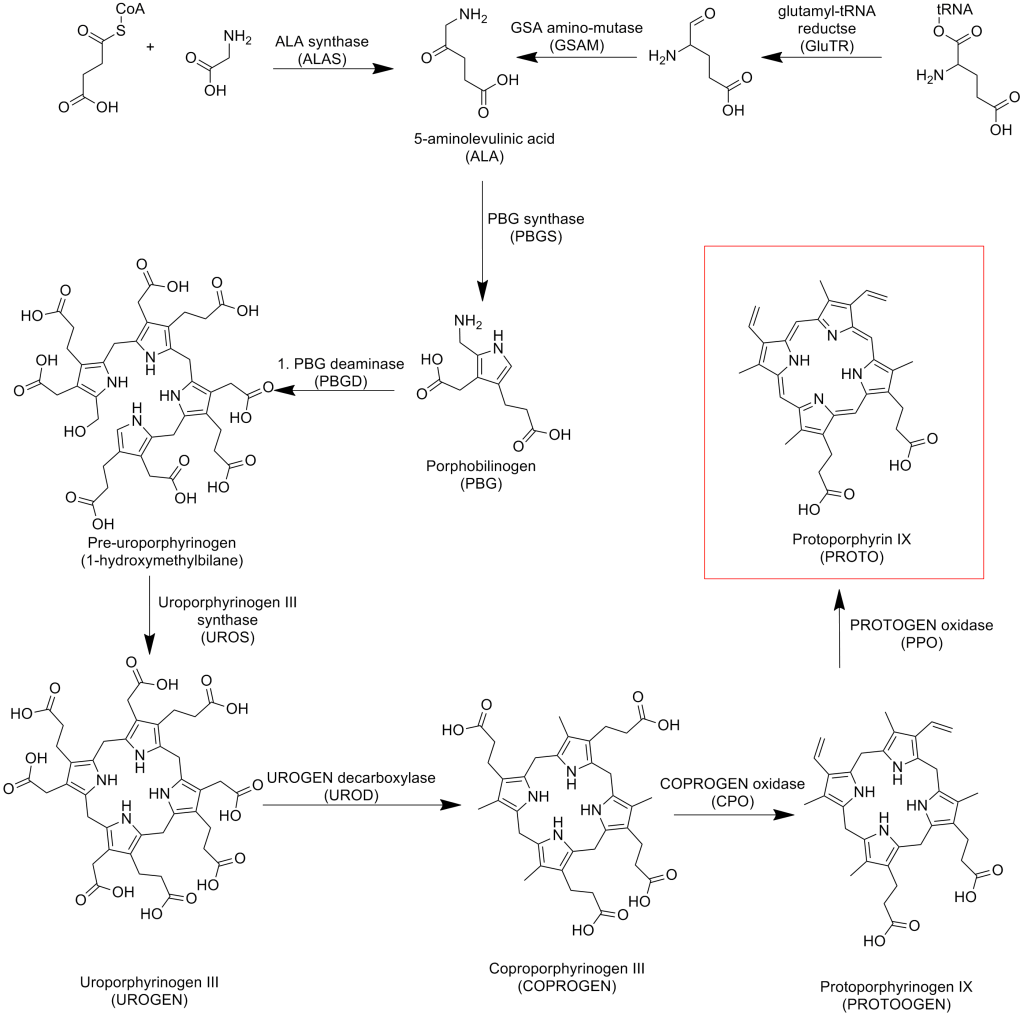
Biosyntese av heme
Produksjonen av heme er en livsviktig prosess som foregår i nesten alle kroppens celler, men spesielt aktiv i leverceller og i de erytropoietiske cellene i benmargen. I leveren er hovedformålet å produsere heme for enzymer som cytokromer, mens i benmargen brukes heme primært til dannelse av hemoglobin i røde blodceller.
Hemesyntesen er en kompleks prosess som krever nøyaktig koordinering av flere trinn. Denne biosyntesen involverer åtte enzymatiske reaksjoner som finner sted både i mitokondriene og i cytoplasma.
Utgangsstoffer – byggesteinene
Syntesen av heme starter med to enkle molekyler:
- Glysin – en aminosyre som finnes i kroppen og maten vi spiser.
- Succinyl-CoA – et mellomprodukt i sitronsyresyklusen.
Disse to molekylene kombineres i en reaksjon som danner grunnlaget for porfyrinringen som senere skal binde jern og danne heme.
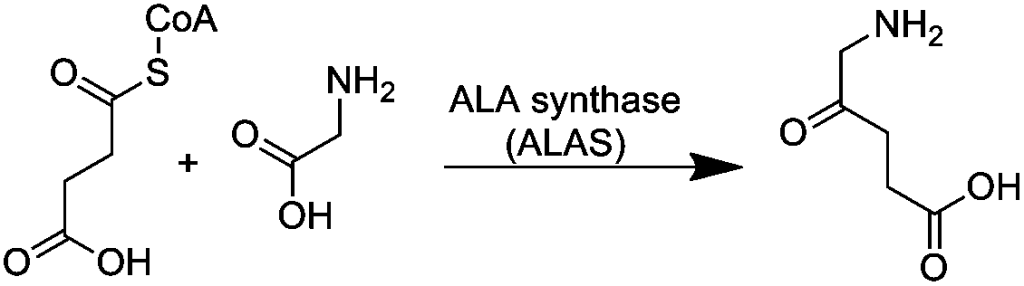
Hemesyntesens første trinn: Dannelsen av ALA
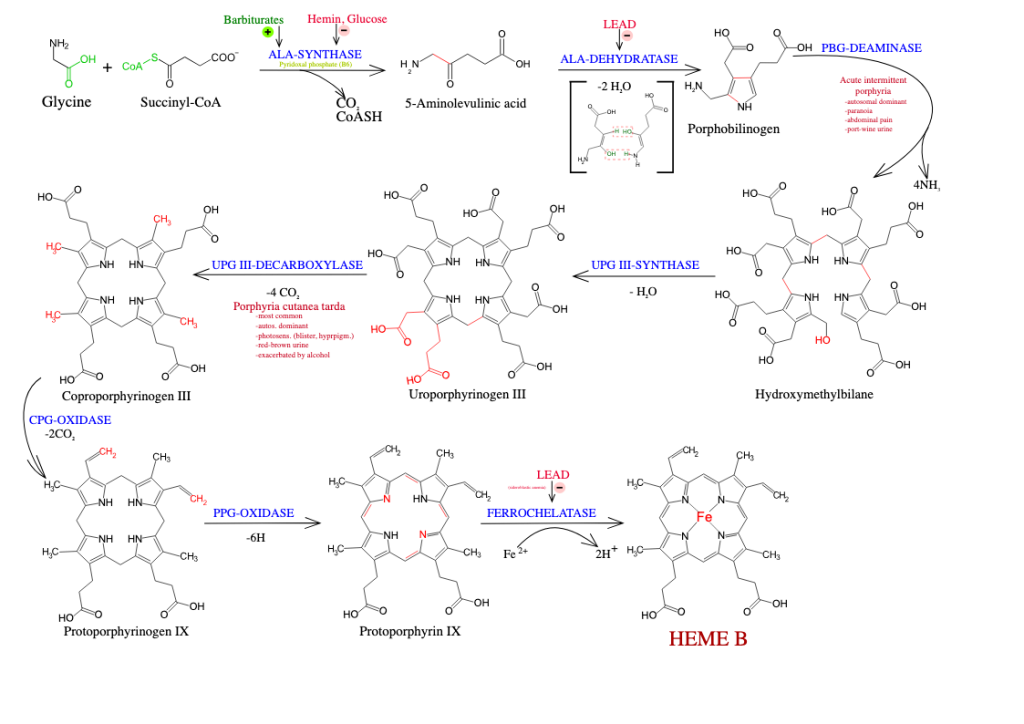
Det første og hastighetsbestemmende steget i hemesyntesen katalyseres av enzymet ALA-syntase (aminolevulinsyre-syntase). I denne reaksjonen smeltes glysin og succinyl-CoA sammen til et nytt molekyl kalt aminolevulinsyre (ALA).
Denne reaksjonen skjer inne i mitokondriene og krever vitamin B6 (i form av pyridoksalfosfat) som nødvendig kofaktor. Fordi dette steget bestemmer tempoet på hele hemesyntesen, er det også her kroppen regulerer produksjonen mest nøye, hovedsakelig ved negativ feedback fra ferdiglaget heme som hemmer ALA-syntase. Mutasjoner eller vitamin B6-mangel kan forstyrre dette steget og føre til sykdommer som sideroblastisk anemi.
De videre syntesetrinnene – fra ALA til porfyrinring
Etter dannelsen av ALA transporteres molekylene ut i cytoplasmaet, hvor en serie enzymatiske reaksjoner bygger opp den karakteristiske porfyrinringen.
Steg 2 i syntesen innebærer at to ALA-molekyler kondenseres sammen til porfobilinogen. Denne reaksjonen katalyseres av enzymet ALA-dehydratase (også kalt porfobilinogen-syntase). Enzymet er sensitivt for tungmetaller som bly, og hemming av ALA-dehydratase er en viktig mekanisme bak blyforgiftning.
Videre, i steg 3, kobles fire porfobilinogen-molekyler sammen for å danne en lineær tetrapyrrol kalt hydroksymetylbilan. Denne reaksjonen katalyseres av enzymet porfobilinogen deaminase (også kjent som uroporfyrinogen I syntase). Feil i dette enzymet kan føre til sykdommen akutt intermitterende porfyri, som karakteriseres av anfall med magesmerter og nevropsykiatriske symptomer.
Når hydroksymetylbilan er dannet, fortsetter syntesen gjennom flere mellomsteg:
- Hydroksymetylbilan omdannes videre til uroporfyrinogen III ved hjelp av uroporfyrinogen III-syntase.
- Uroporfyrinogen III modifiseres til koproporfyrinogen III via uroporfyrinogen dekarboksylase.
Alle disse trinnene skjer i cytoplasmaet, og mellomproduktene transporteres deretter tilbake inn i mitokondriene for ferdigstilling.
De siste trinnene – ferdigstillelse av heme
Inne i mitokondriene fortsetter prosessen:
- Koproporfyrinogen III omdannes til protoporfyrinogen IX av enzymet koproporfyrinogen oksidase.
- Protoporfyrinogen IX oksideres videre til protoporfyrin IX ved hjelp av protoporfyrinogen oksidase.
- Til slutt settes et jernatom (Fe²⁺) inn i midten av protoporfyrin IX-ringen. Dette skjer ved hjelp av enzymet ferrokelatase, som dermed fullfører dannelsen av funksjonelt heme. Hemming av ferrokelatase, for eksempel ved blyforgiftning, kan føre til akkumulering av forløpermolekyler og hemme effektiv hemesyntese.
Hvor skjer hemesyntesen?
- I benmargen: Omtrent 85 % av kroppens hemeproduksjon skjer her, der heme brukes til å lage hemoglobin for nye røde blodceller.
- I leveren: Leveren produserer heme primært for sine egne behov, som i produksjonen av cytokrom P450-enzymer som brukes i kroppens avgiftningssystemer.
Syntesens plassering i cellen
- De første trinnene (som dannelsen av ALA) og de siste trinnene (innsetting av jern) foregår i mitokondriene.
- Midtfasen, hvor porfyrinmellomproduktene dannes og modifiseres, skjer i cytoplasmaet.
Denne spesifikke fordelingen mellom mitokondrier og cytoplasma sikrer effektiv koordinering, nøye regulering og beskyttelse mot toksiske mellomprodukter underveis i hemesyntesen.
De siste trinnene – ferdigstillelse av heme
I mitokondriene omdannes koproporfyrinogen til protoporfyrinogen, som videre oksideres til protoporfyrin IX – den siste ringen før heme dannes.
Til slutt settes et jernatom (Fe²⁺) inn i midten av porfyrinringen ved hjelp av enzymet ferrokelatase. Dette trinnet fullfører dannelsen av funksjonelt heme.
Hvor skjer hemesyntesen?
- I benmargen: Omtrent 85 % av kroppens hemeproduksjon skjer her, der heme brukes til å lage hemoglobin for nye røde blodceller.
- I leveren: Leveren produserer heme primært for sine egne behov, som i produksjonen av cytokrom P450-enzymer for avgiftning.
Syntesens plassering i cellen
- De første og siste trinnene foregår i mitokondriene.
- Midtfasen, inkludert dannelsen av porfyrinmellomproduktene, skjer i cytoplasmaet.
Denne spesifikke fordelingen mellom mitokondrier og cytoplasma sikrer effektiv koordinering og kontroll av hemesyntesen.
Regulering av hemesyntesen
Produksjonen av heme må balanseres nøye. For lite heme vil svekke cellens evne til å transportere oksygen og utføre livsviktige kjemiske prosesser, mens for mye fritt heme kan være giftig og skade cellene. Derfor har kroppen utviklet presise mekanismer for å regulere hemesyntesen, både i leveren og i benmargen.
Hovedreguleringen – ALAS-enzymet
Det viktigste kontrollpunktet i hemesyntesen er enzymet ALA-syntase (aminolevulinsyre-syntase), som katalyserer det første trinnet i syntesen: sammensmeltningen av glysin og succinyl-CoA til ALA.
Det finnes to hovedtyper av ALA-syntase:
- ALAS1: Uttrykkes i alle vev, men særlig viktig i leverceller.
- ALAS2: Uttrykkes spesifikt i erytropoietiske celler i benmargen.
Begge typene er strengt regulert, men på litt forskjellige måter tilpasset vevets behov.
{kind=link}
Regulering i leveren – respons på heme-nivåer og ytre påvirkninger
I leveren reguleres ALAS1 hovedsakelig gjennom feedback fra heme selv:
Når heme-nivåene er høye, hemmes syntesen av nytt heme. Heme virker ved å:
- Hemme transkripsjonen av ALAS1-genet (slik at mindre ALAS1 produseres).
- Hemme transporten av ALAS1-proteinet inn i mitokondriene.
- Øke nedbrytningen av ALAS1-enzymet som allerede er laget.
Dette betyr at heme fungerer som et negativt feedback-signal som holder produksjonen i sjakk.
I tillegg kan flere ytre faktorer påvirke ALAS1 i leveren:
- Faste og sult kan oppregulere ALAS1, fordi kroppen øker sin egen produksjon av enzymer for å håndtere endrede metabolske behov.
- Legemidler som stimulerer cytokrom P450-systemet, for eksempel barbiturater, øker behovet for heme i leveren og kan derfor også oppregulere ALAS1.
Regulering i benmargen – hemesyntesen avhenger av jern
I de erytropoietiske cellene i benmargen, som lager røde blodceller, er hemesyntesen nøye regulert etter hvor mye jern som er tilgjengelig. Det er fordi kroppen ikke vil produsere heme hvis det ikke finnes jern til å fullføre molekylet.
Et viktig kontrollpunkt er enzymet ALAS2 (aminolevulinsyre-syntase 2), som er spesifikt for røde blodcelleforløpere. ALAS2-produksjonen styres på mRNA-nivå via noe som kalles et jernrespons-element (IRE) – en liten sekvens i starten av ALAS2-mRNA.
Dette systemet fungerer slik:
- Når det er lite jern, er et protein kalt IRP (Iron Regulatory Protein) aktivt og binder seg til IRE på ALAS2-mRNA.
Denne bindingen blokkerer translasjonen, slik at ALAS2 ikke blir produsert.
Resultat: hemesyntesen settes på vent. - Når det er god tilgang på jern, binder jern seg til IRP.
Dette gjør at IRP slipper taket i mRNA.
Da blir ALAS2-mRNA tilgjengelig for ribosomer, og enzymet kan produseres.
Resultat: ALAS2 øker, og heme produseres i takt med at cellen lager hemoglobin.
Med andre ord:
🔻 Lite jern → IRP binder IRE → ALAS2 blokkeres → mindre heme
🔺 Mye jern → IRP slipper IRE → ALAS2 uttrykkes → mer heme
Denne mekanismen sørger for at produksjonen av heme alltid er tilpasset tilgjengeligheten av jern – og hindrer at cellen kaster bort ressurser på å lage halvferdige molekyler. Også viktig å både vite og huske at mange av mellomproduktene i dannelse av heme er giftige.
Denne koblingen mellom jern og hemesyntese sikrer at røde blodceller ikke produserer ubrukelige globinkjeder uten heme, som ville vært både energikrevende og skadelig.
Klinisk side – porfyrisykdommer
Forstyrrelser i hemesyntesen kan føre til en gruppe sjeldne, men alvorlige tilstander kjent som porfyrisykdommer. Disse skyldes som regel arvelige mutasjoner i ett av enzymene som inngår i synteseveien for heme. Når et enzym ikke fungerer som det skal, vil mellomprodukter i syntesen hope seg opp – og det er disse opphopede stoffene som gir symptomer.
Symptombildet varierer med hvilket enzym som er rammet og hvilke mellomprodukter som hoper seg opp:
- Noen former gir alvorlige magesmerter, kvalme og nevrologiske symptomer som angst, forvirring og muskelsvakhet – dette er typisk for de akutte porfyrier, hvor vannløselige forløpere som ALA og porfobilinogen akkumulerer og påvirker nervesystemet.
- Andre former gir hovedsakelig lysømfintlighet, der huden reagerer kraftig på sollys – dette skjer når porfyriner som er lysaktive hoper seg opp i huden.
I leveren reguleres hemesyntesen via enzymet ALAS1, som normalt holdes i sjakk av ferdiglaget heme (negativ feedback). Men visse faktorer – som faste, alkohol og enkelte legemidler – kan øke ALAS1-aktiviteten. Dette skjer fordi leveren trenger flere cytokromer (hemeholdige enzymer) for avgiftning under slike forhold. Når ALAS1 øker, øker også gjennomstrømmingen i synteseveien – og hvis et enzym i kjeden er defekt, fører det til økt produksjon og opphopning av toksiske mellomprodukter.
Derfor er det kritisk å unngå utløsende faktorer hos personer med kjent porfyri. Behandlingen fokuserer på å redusere ALAS1-aktiviteten, for eksempel med glukose eller hemin, som begge hemmer ALAS1 og reduserer syntesen av de skadelige stoffene.
Nedbrytning av heme
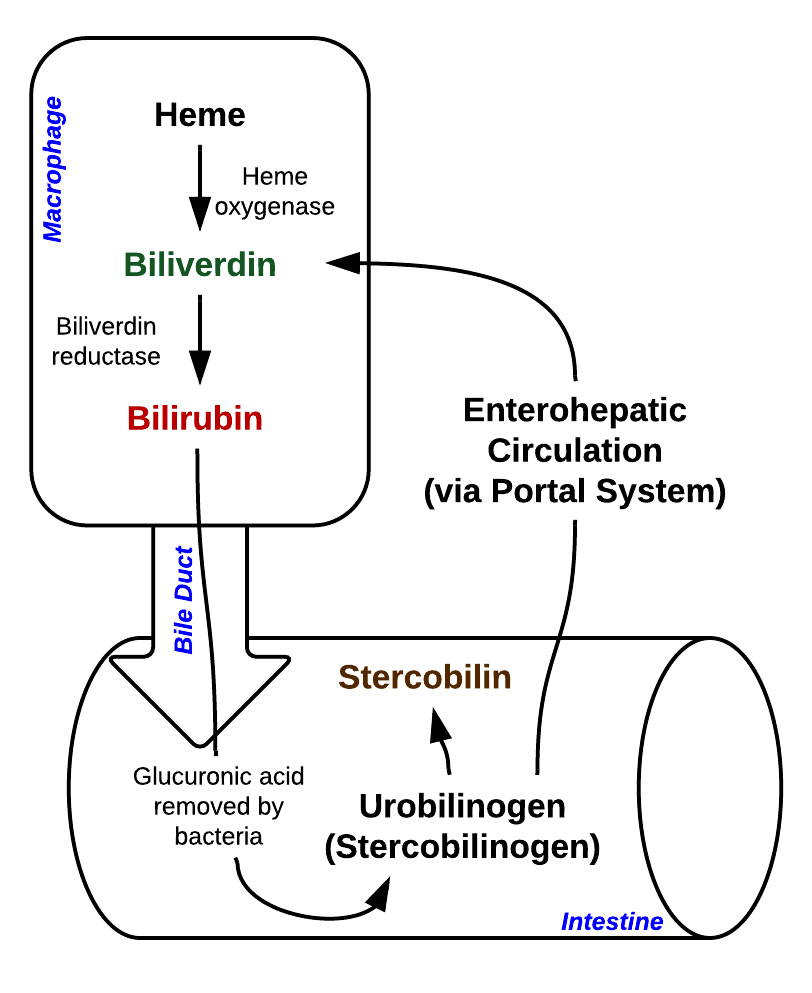
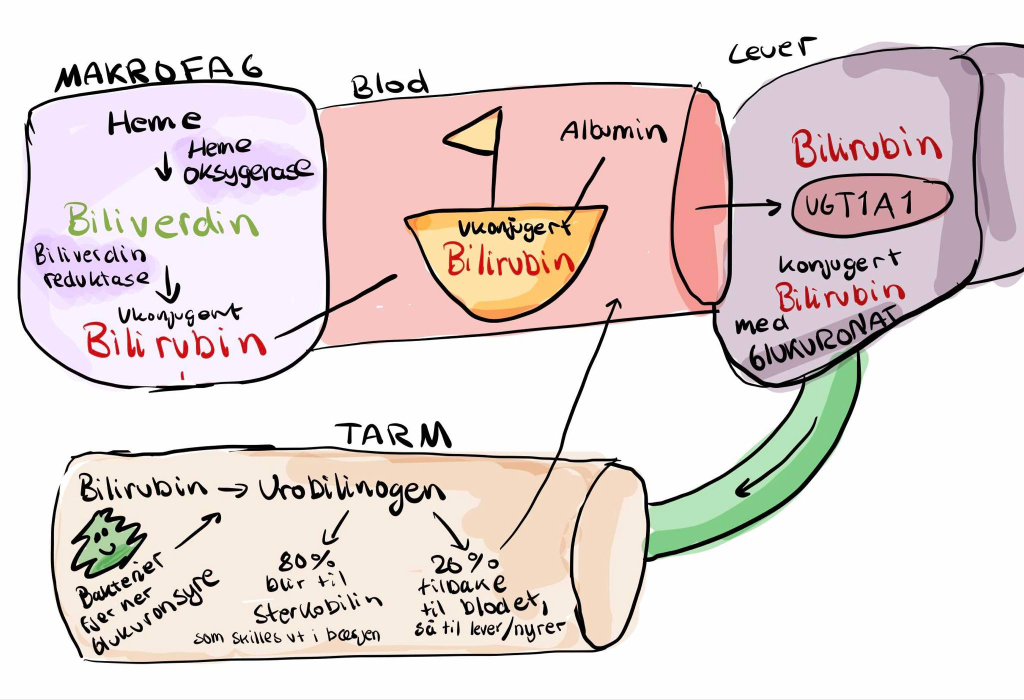
Etter å ha tjent sin livsviktige funksjon i hemoglobin og andre hemeproteiner, må heme brytes ned på en kontrollert måte. Nedbrytningen er helt avgjørende for å beskytte kroppen mot skade, fordi fritt heme og jern kan være svært reaktive og fremme dannelse av skadelige frie oksygenforbindelser. Nedbrytningen av heme starter når gamle eller skadde røde blodceller fjernes fra sirkulasjonen.
Fjerning av gamle erytrocytter
Gamle erytrocytter har en levetid på omtrent 120 dager før de gjenkjennes og fjernes av makrofager i det retikuloendoteliale systemet (RES). RES består primært av makrofager i milten, leveren og benmargen. Disse makrofagene fungerer som kroppens «oppryddingsarbeidere» og starter prosessen med å bryte ned hemoglobin.
Trinnene i nedbrytningen av hemoglobin
Når makrofager tar opp gamle erytrocytter, brytes hemoglobin ned i sine tre hovedkomponenter:
- Globin: Globinkjedene brytes ned til sine bestanddeler, aminosyrer, som enten kan gjenbrukes eller brukes som energikilde.
- Jern: Jernet fra hemegruppen resirkuleres. Det fraktes ut av makrofagen bundet til transportproteinet transferrin og kan lagres i ferritin eller brukes til å lage nye erytrocytter i benmargen.
- Heme: Selve hemegruppen brytes videre ned i en spesialisert prosess som håndteres av enzymet heme oxygenase.
Omdannelse av heme til bilirubin
- Heme oxygenase: Dette enzymet katalyserer det første trinnet i hemenedbrytningen. Det åpner porfyrinringen ved å spalte heme til biliverdin, samtidig som det frigjøres jern (Fe²⁺) og karbonmonoksid (CO).
Karbonmonoksid fungerer her som et signalmolekyl, men i større mengder kan det være toksisk. - Biliverdin reduktase: Biliverdin, som er grønt, reduseres raskt til bilirubin, som har en gul-oransje farge. Denne reaksjonen gjør bilirubin mer fettløselig, men samtidig vanskelig å transportere i vannbasert blodplasma.
Transport av ukonjugert bilirubin
Fordi ukonjugert bilirubin er fettløselig og potensielt giftig, må det transporteres bundet til albumin i blodet. Albumin fungerer som en beskyttende «fraktbåt» som sørger for at bilirubin ikke skader cellene på vei til leveren, hvor det videre skal behandles.
På dette tidspunktet er bilirubin ukonjugert – det vil si at det ennå ikke er gjort vannløselig – og kan ikke skilles ut av kroppen før det er modifisert i leveren.
Transport og behandling av bilirubin i leveren
Når bilirubin er dannet i makrofagene og bundet til albumin i blodbanen, transporteres det til leveren for videre behandling. Leveren fungerer her som en sentral behandlingsstasjon som gjør bilirubin vannløselig slik at det kan skilles ut av kroppen på en trygg måte.
Opptak av bilirubin i leveren
Ved ankomst til leveren frigjøres bilirubin fra albumin og tas aktivt opp av hepatocyttene (levercellene). Transporten over cellemembranen skjer via spesifikke transportproteiner som effektivt overfører bilirubin fra blodet og inn i cellens cytoplasma.
Konjugering av bilirubin – vannløselig form
Inne i hepatocyttene undergår bilirubin en kjemisk modifikasjon som kalles konjugering. Denne prosessen katalyseres av enzymet UDP-glukuronosyltransferase (UGT1A1), som kobler to glukuronsyremolekyler til bilirubin.
Resultatet er konjugert bilirubin, også kalt bilirubin-diglukuronid.
Konjugert bilirubin er:
- Vannløselig (kan nå fraktes i kroppens væsker uten albumin).
- Ikke-giftig.
- Klar for aktiv utskillelse i galle.
Transport av konjugert bilirubin til gallegangene
Etter konjugeringen transporteres bilirubin ut av hepatocyttene via spesifikke ATP-drevne transportproteiner, blant annet MRP2 (Multidrug Resistance-associated Protein 2).
Konjugert bilirubin pumpes aktivt inn i gallekanalikuli – små kanaler som leder gallen fra hepatocyttene og samler den opp i større galleveier.
Gallen, som nå inneholder konjugert bilirubin sammen med gallesalter, kolesterol og avfallsstoffer, føres mot galleblæren for lagring eller direkte til tarmen via gallegangen.
Betydningen av konjugering
Konjugeringen av bilirubin er en kritisk beskyttelsesmekanisme:
- Den gjør at bilirubin kan utskilles effektivt via galle og tarm.
- Den hindrer at fettløselig, ukonjugert bilirubin akkumuleres i blodet og forårsaker skade.
- Den sikrer at bilirubin, som i utgangspunktet er potensielt giftig, håndteres på en trygg måte uten å skade lever eller andre vev.
Ved svikt i denne prosessen, enten på grunn av genetiske defekter (som ved Gilbert syndrom) eller leversykdommer, kan ukonjugert bilirubin hope seg opp i blodet og føre til utvikling av gulsott (ikterus).
Utskillelse og videre omdanning av bilirubin i tarmen
Etter å ha blitt konjugert og pakket inn i gallen, transporteres bilirubin fra leveren gjennom gallegangene og tømmes ut i tarmen, nærmere bestemt i duodenum. Her fortsetter bilirubin sin ferd gjennom fordøyelseskanalen, men reisen er langt fra slutt.
Bilirubin i tarmen – møte med bakterier
I tynntarmen og særlig i tykktarmen møter bilirubin kroppens normale tarmflora – de mange bakteriene som lever naturlig i fordøyelsessystemet. Disse bakteriene har evnen til å omdanne konjugert bilirubin videre.
Enzymer fra bakteriene spalter av glukuronsyren som ble påkoblet i leveren, og bilirubin omdannes til en fargeløs forbindelse kalt urobilinogen.
Hva skjer videre med urobilinogen?
Etter dannelsen av urobilinogen finnes det flere mulige veier videre:
- Hoveddelen (~80–90 %) forblir i tarmen og oksideres videre av bakteriene til stercobilin.
Stercobilin gir den karakteristiske brunfargen til avføringen. - En mindre del (~10–20 %) av urobilinogen reabsorberes tilbake til blodet gjennom tarmveggen.
Det som reabsorberes følger to mulige skjebner:- En liten mengde resirkuleres tilbake til leveren via det enterohepatiske kretsløpet.
- Resten transporteres til nyrene, hvor det oksideres til urobilin og skilles ut med urinen, noe som gir urin sin gule farge.
Viktige poenger om utskillelsen
- Konjugert bilirubin gjør det mulig å kvitte seg med avfall fra nedbrutt heme på en trygg måte gjennom gallen.
- Bakterienes omdanning av bilirubin i tarmen er avgjørende for den normale fargen på både avføring og urin.
- Eventuelle forstyrrelser i denne prosessen, for eksempel blokkering av galleganger, kan føre til misfarging av både urin (mørk) og avføring (lys/grå), og utvikling av ikterus.
Klinisk relevans – Ikterus og hyperbilirubinemi
Små forstyrrelser i metabolismen av heme og håndteringen av bilirubin kan gi synlige og alvorlige konsekvenser. Den mest kjente manifestasjonen er ikterus, også kalt gulsott, hvor gulfarging av hud og øyne oppstår på grunn av økte nivåer av bilirubin i blodet. For å forstå hvordan ikterus utvikler seg, må vi se på hvor i prosessen forstyrrelsene skjer.
Hva er ikterus og hyperbilirubinemi?
Ikterus betegner gulfarging av kroppens vev, og skyldes akkumulering av bilirubin i blod og vev. Normalt holdes bilirubinnivået i blodet lavt, men når produksjonen øker, leverens behandling svekkes, eller utskillelsen hindres, vil bilirubinet hope seg opp. Når konsentrasjonen i blodet overstiger omtrent 20–25 mikromol per liter, blir gulfargen først synlig i sclerae (det hvite i øynene), og senere i hud og slimhinner.
Hyperbilirubinemi er den biokjemiske betegnelsen på forhøyede bilirubinnivåer i blodet, og kan deles i to hovedformer: ukonjugert hyperbilirubinemi, der det er overskudd av fettløselig, ukonjugert bilirubin, og konjugert hyperbilirubinemi, der vannløselig, konjugert bilirubin dominerer. Å skille mellom disse formene gir viktig informasjon om hvor i bilirubinmetabolismen problemet ligger.
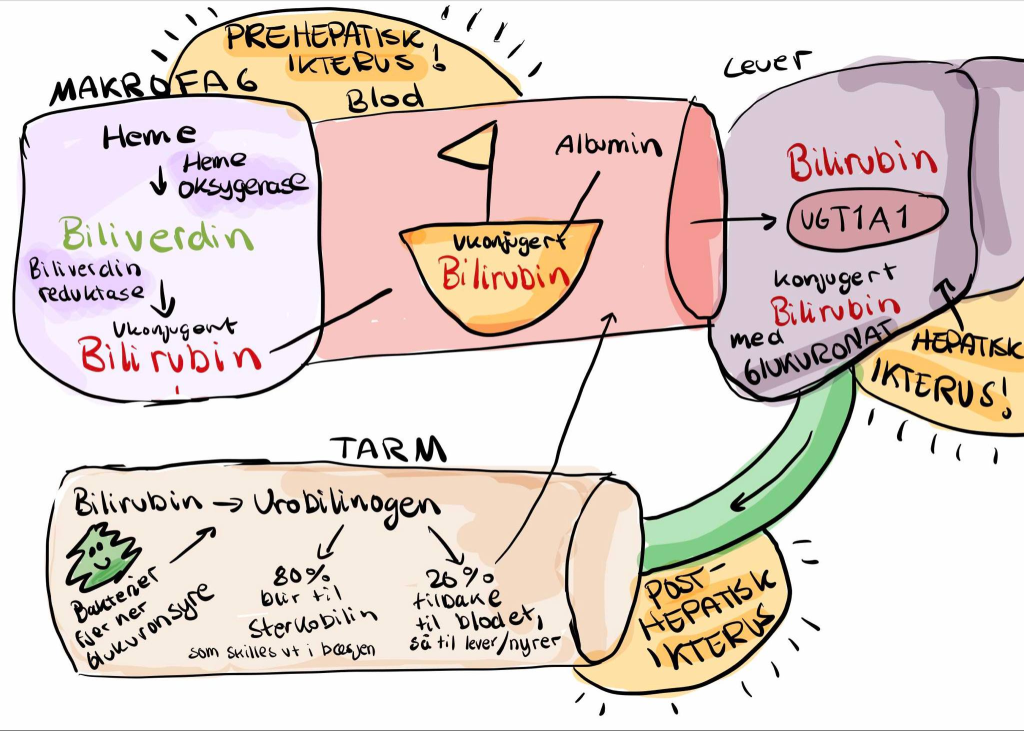
Tre hovedårsaker til ikterus
For å systematisere årsakene til ikterus deler vi dem inn etter hvor forstyrrelsen oppstår: før leveren (prehepatisk), i leveren (hepatisk) eller etter leveren (posthepatisk).
Ved prehepatisk ikterus skyldes hyperbilirubinemi en overproduksjon av bilirubin. Dette skjer når røde blodceller ødelegges i stort antall, som ved hemolytiske anemier eller ved store hematomer som resorberes. Leveren rekker ikke å konjugere alt bilirubinet som produseres, og resultatet blir en økning av ukonjugert bilirubin i blodet.
Hepatisk ikterus oppstår når selve levercellene er skadet og ikke klarer å ta opp, konjugere eller skille ut bilirubin effektivt. Dette kan være forårsaket av infeksjoner som virushepatitt, toksisk skade ved alkoholbruk eller avansert leversykdom som cirrhose. Fordi både opptak, konjugering og utskillelse kan rammes, sees ofte en blanding av ukonjugert og konjugert hyperbilirubinemi.
Ved posthepatisk ikterus er det ikke produksjonen av bilirubin eller leverens evne til å konjugere som svikter, men selve transporten av bilirubin ut av leveren. Galleutløpet kan være blokkert, for eksempel av gallestein, svulster eller betennelse. Fordi bilirubinet allerede er konjugert, fører en slik blokkering til økt konjugert bilirubin i blodet.
Kliniske kjennetegn
De kliniske tegnene på ikterus varierer avhengig av hvor forstyrrelsen sitter. Ved prehepatisk ikterus vil avføringen og urinen som regel være normale i farge, fordi leverens utskillelse fungerer normalt. Ved hepatisk ikterus kan både urinen bli mørk og avføringen lys, avhengig av graden av leverskade. I posthepatisk ikterus vil urinen bli mørk, fordi konjugert bilirubin skilles ut via nyrene, og avføringen kan bli blek eller grå, på grunn av mangel på sterkobilin som normalt gir avføringen dens brune farge. Kløe er også vanlig ved posthepatisk ikterus, som følge av opphopning av gallesyrer i blodet.
Hvorfor endrer urin og avføring farge?
Ved normal utskillelse skilles bilirubin ut i tarmen og omdannes til sterkobilin, som gir brun farge til avføringen. Når gallegangene er blokkert, hindres denne prosessen. I stedet reabsorberes konjugert bilirubin til blodet og skilles ut i urinen, hvor det gir en mørk gulfarge. Den manglende sterkobilinproduksjonen gjør samtidig at avføringen blir blek.
Oppsummering
Ikterus og hyperbilirubinemi gir en viktig klinisk pekepinn på hvor i metabolismen av heme og bilirubin prosessen svikter. Å forstå om det dreier seg om en prehepatisk, hepatisk eller posthepatisk årsak er essensielt for å finne riktig diagnose og behandling.
📚 Anki-kort
Obs, tomt! Kommer etterhvert <3
📝 Eksamensoppgaver
Obs, tomt! Kommer etterhvert <3
👨⚕️ Klinisk case
Obs, tomt! Kommer etterhvert <3
❓ Test deg selv
Obs, tomt! Kommer etterhvert <3